Physical model induction with QuanSA™: Affinity prediction that is synergistic with simulation-based methods
The QuanSA method To define a ‘pocket field’, an initial alignment of all training molecules is constructed and function parameters…
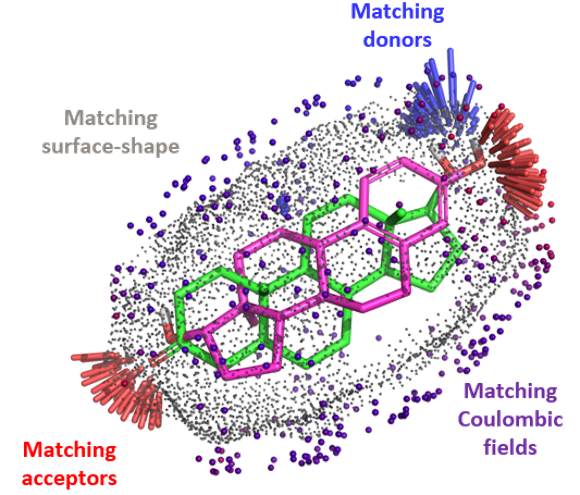
The QuanSA method To define a ‘pocket field’, an initial alignment of all training molecules is constructed and function parameters…
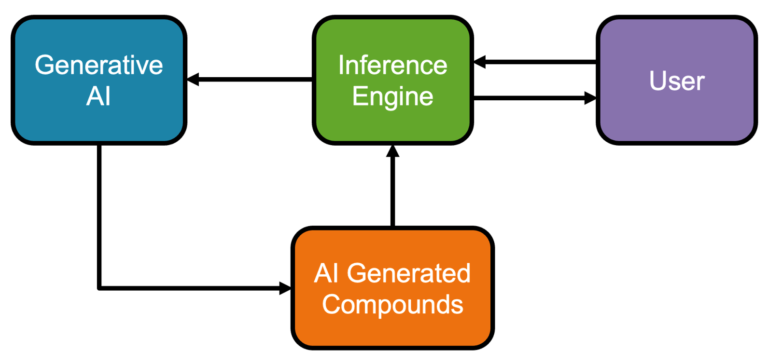
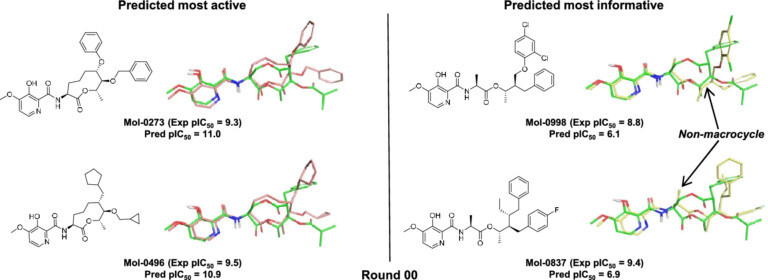
Pairing AI with human expertise We present a novel AI compound optimisation system, designed to include human oversight as a…
Introduction Using the integrated set of computational methods within the BioPharmics™ Platform, macrocycles can be effectively modelled for lead optimisation.…

Introduction The emergence of resistance and increased stringency of regulatory requirements have created a need for new agrochemicals. The long…
In this paper, we describe an extended benchmark for non-cognate docking of macrocyclic ligands, and the superior performance of Surflex-Dock…
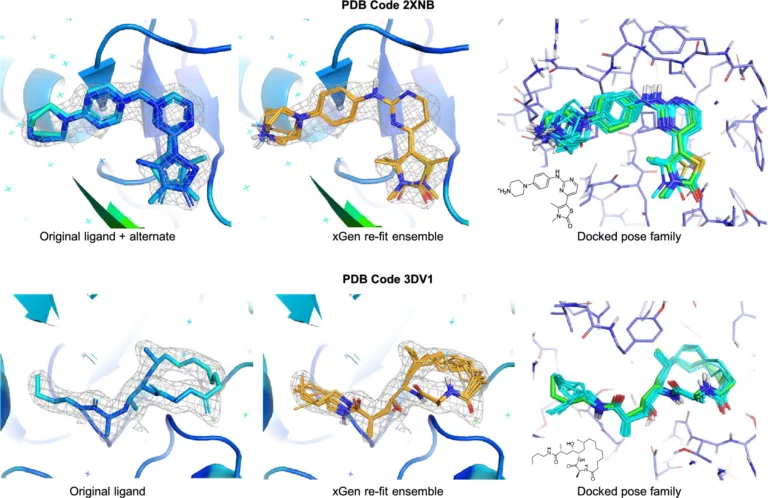
Scaffold replacement as part of an optimisation process that requires maintenance of potency, desirable biodistribution, metabolic stability, and considerations of synthesis at very large scale is a complex challenge.
Predicting metabolism at an early stage is important in maximising the chance of a drug’s success. However, accurate, useful models…
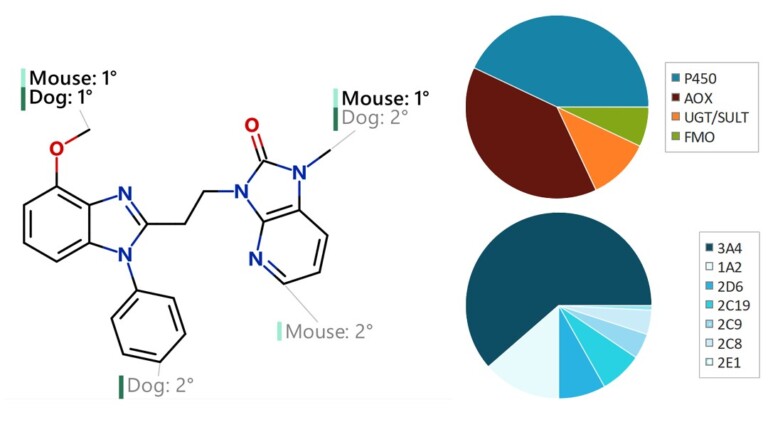
This peer-reviewed paper in Xenobiotica describes a new method to determine the most likely experimentally-observed routes of metabolism and metabolites based on our WhichP450™, regioselectivity and new WhichEnzyme™ model.
Systematic optimisation of large macrocyclic peptide ligands is a serious challenge. Here, we describe an approach for lead optimisation using the PD-1/PD-L1 system as a retrospective example of moving from initial lead compound to clinical candidate.
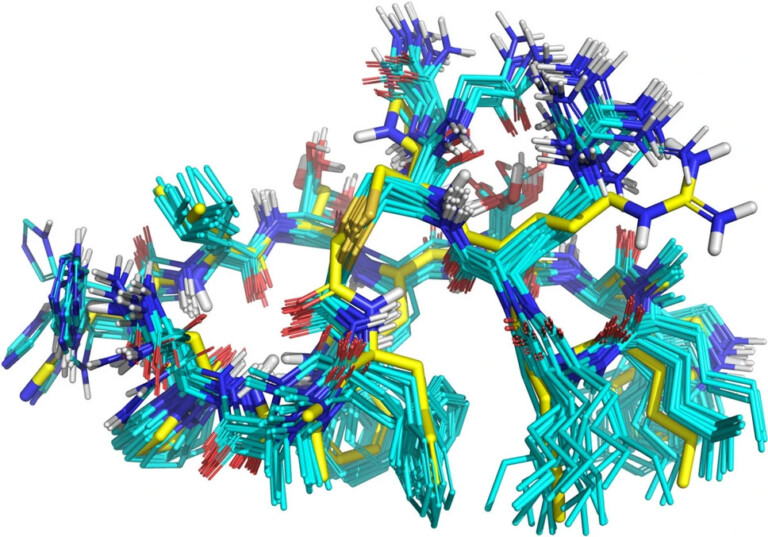
The solution structure of the minor conformer of rapamycin was investigated using a combination of NMR techniques and computational methods

This paper describes a model to predict whether a particular site on a molecule will be metabolised by cytosolic sulfotransferase enzymes (SULTs).
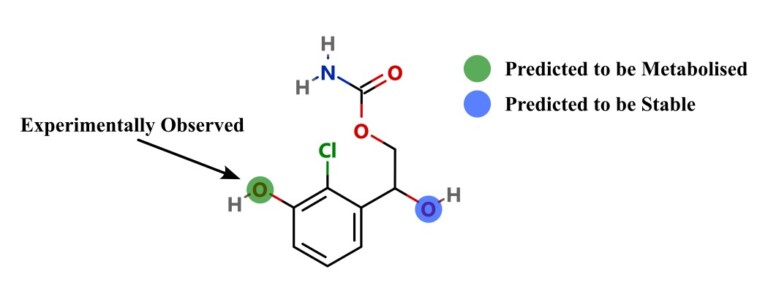
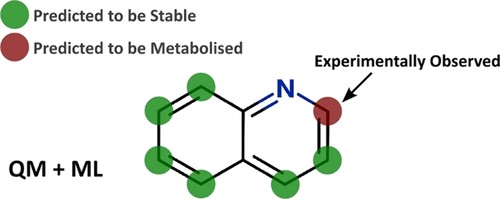
Introduction Predicting sites of metabolism (SoM) enable chemists to be more efficient in optimising the structure of new chemical entities…
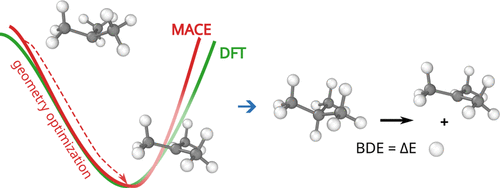
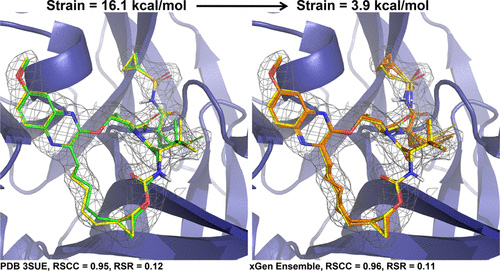
We show that the distribution of expected global strain energy values is dependent on molecular size in a superlinear manner. The distribution of strain energy follows a rectified normal distribution whose mean and variance are related to conformational complexity.
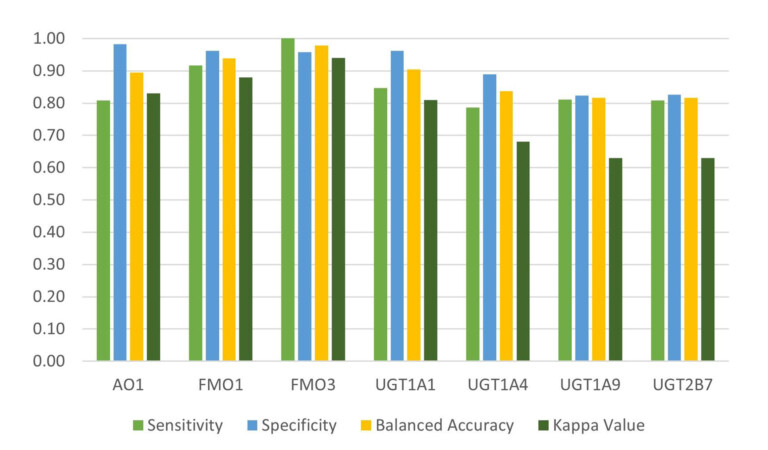
This paper describes the prediction of the regioselectivity of metabolism by AOs, FMOs and UGTs for humans and CYPs for three preclinical species.
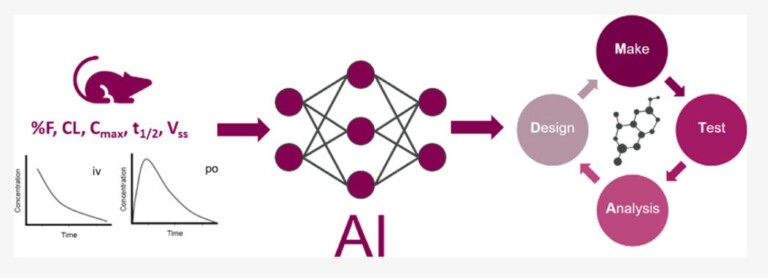
This article is a collaboration with Intellegens, the University of Cambridge and AstraZeneca. It provides a proof-of-concept study in which Cerella™ is used to predict rat in vivo pharmacokinetic (PK) parameters and concentration–time PK profiles.
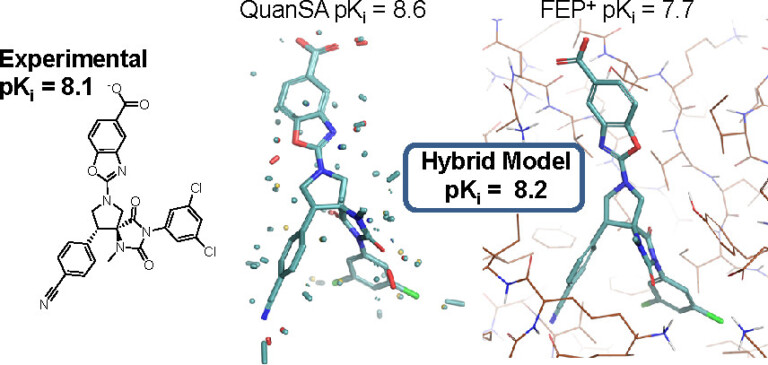
We present results on the extent to which physics-based simulation (exemplified by FEP+) and focused machine learning (exemplified by QuanSA) are complementary for ligand affinity prediction.
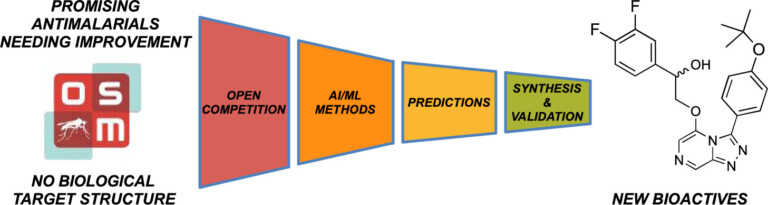
In this study, we identified a new antimalarial with an unusual structure – the only compound in the competition to be proven active, opening up new chemistry for exploration.
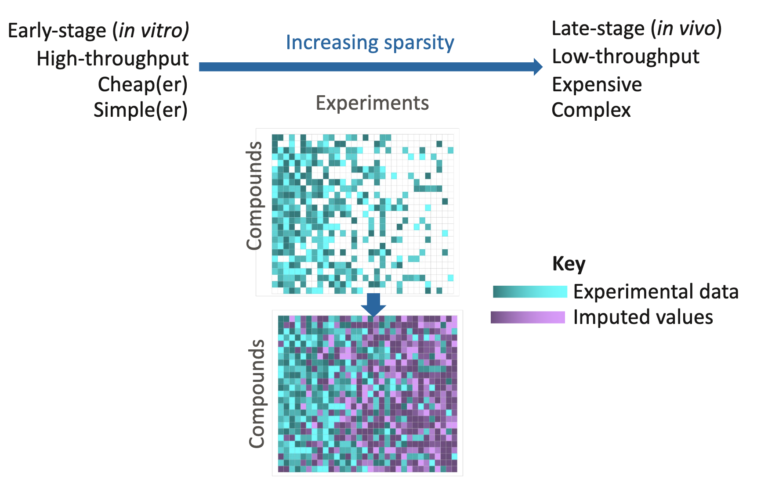
In this article, the team demonstrates the application of Alchemite™, a deep learning imputation method which underpins our Cerella™ technology, to physicochemical and sensory data.
OA paper outlining the practical applications of deep imputation on large-scale drug discovery data. It compares deep learning to traditional QSAR methods.
