Is AI improving drug discovery?
Is AI-guided drug discovery faster and cheaper? The evidence for this is, by definition, anecdotal. No one runs the same…

Is AI-guided drug discovery faster and cheaper? The evidence for this is, by definition, anecdotal. No one runs the same…
What’s the purpose of a predictive model? What’s the value of predictive models for drug discovery? Most of the undergraduate…

The role of generative chemistry in drug discovery A key difficulty in finding new drugs is the sheer size of…

Discover which metabolite prediction software is best for your needs in this comprehensive guide from Optibrium. Compare top tools like Meteor Nexus, MetaSite, and StarDrop to make informed decisions for drug metabolism prediction

We recently published a case study with Amazon Web Services, detailing how we were able to scale our StarDrop platform…

The joint ISSX/JSSX meeting is for researchers looking to gain a deeper understanding of drug metabolism and pharmacokinetics.

Join Optibrium’s Chris Khoury at the 38th NMCS meeting in Seattle, 23-26 June
Nearly all computational methods in the CADD field depend on parameters whose values are derived from various types of experimental…

Develop advanced MPO strategies and target the right compounds, faster.
We’re diving back into our favourite subject: multi-parameter optimisation.

Everyone knows smooth collaboration can speed up successful drug discovery projects. But how can we collaborate easily in drug discovery…

Successful drugs require a delicate balance of many properties, such as potency, ADME and toxicity, to meet a project’s therapeutic objective. To make decisions about compound progression and assay selection, the available data must be assessed against project-specific criteria. However, the data on which we base our decisions often come from different sources and can vary in quality, so how can we use this information to make confident decisions? In addition, how can we be sure that the criteria we’re using are the most appropriate?

Interpreting metabolite-ID experiments; determining the right species for animal studies; providing optimisation suggestions for your medicinal chemistry colleagues to overcome…
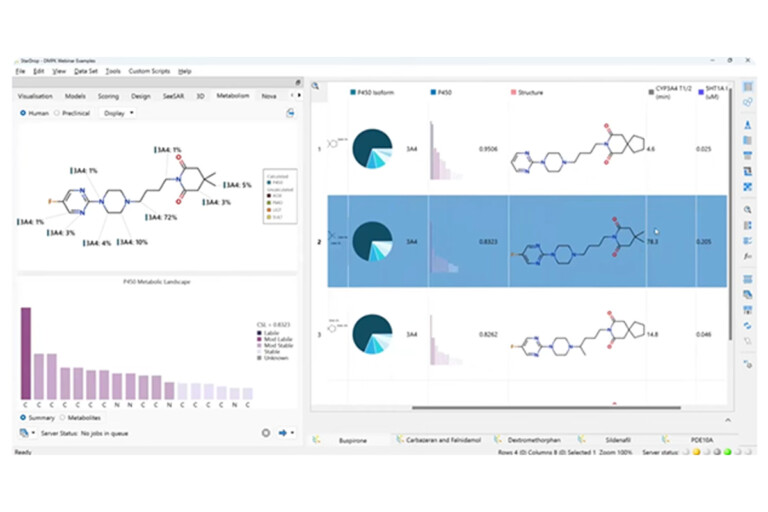
Predicting metabolism at an early stage is important in maximising the chance of a drug’s success. However, accurate, useful models…
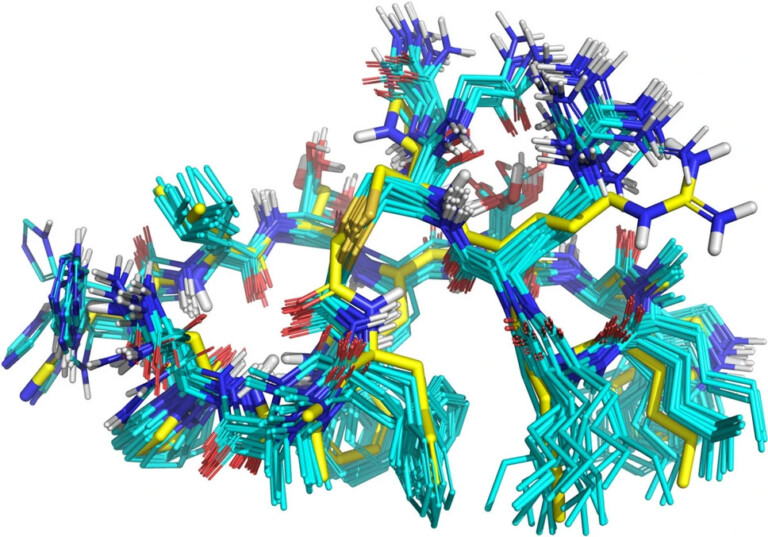
Systematic optimisation of large macrocyclic peptide ligands is a serious challenge. Here, we describe an approach for lead optimisation using the PD-1/PD-L1 system as a retrospective example of moving from initial lead compound to clinical candidate.
Watch our panel of experts as they discuss tactics to achieve success in your medicinal chemistry projects, experiences they’ve had and advice they would give. Learn about the key challenges today’s drug hunter needs to overcome, the skills it takes to gain success across pharma and biotech, and what the future may hold for this industry.

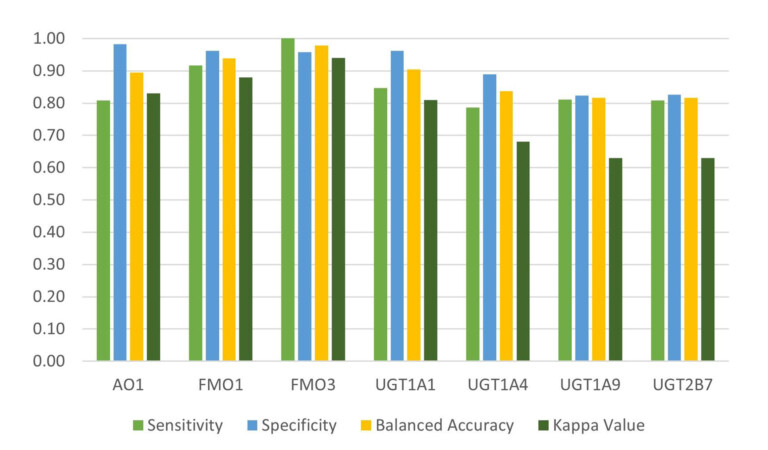
This paper describes a model to predict whether a particular site on a molecule will be metabolised by cytosolic sulfotransferase enzymes (SULTs).
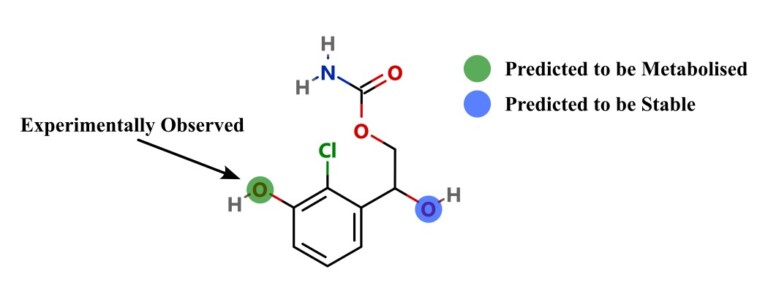
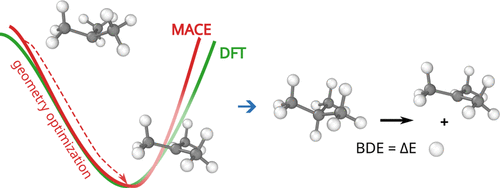
Watch Optibrium CEO Matt Segall and Principal Scientist Mario Öeren as they explore groundbreaking new quantum mechanics and machine learning models which go beyond P450s and provide insights on a broad range of enzymes involved in drug metabolism.
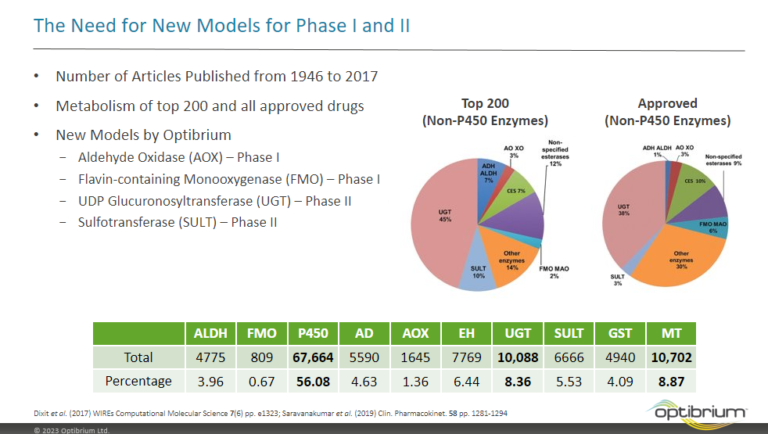
Introduction Predicting sites of metabolism (SoM) enable chemists to be more efficient in optimising the structure of new chemical entities…