38th ACS National Medicinal Chemistry Symposium
Join Optibrium’s Chris Khoury at the 38th NMCS meeting in Seattle, 23-26 June
Join Optibrium’s Chris Khoury at the 38th NMCS meeting in Seattle, 23-26 June
Peer-reviewed study published in Xenobiotica describes an innovative new method that predicts the routes and products of Phase I and II metabolism with high sensitivity and greater precision than
other approaches

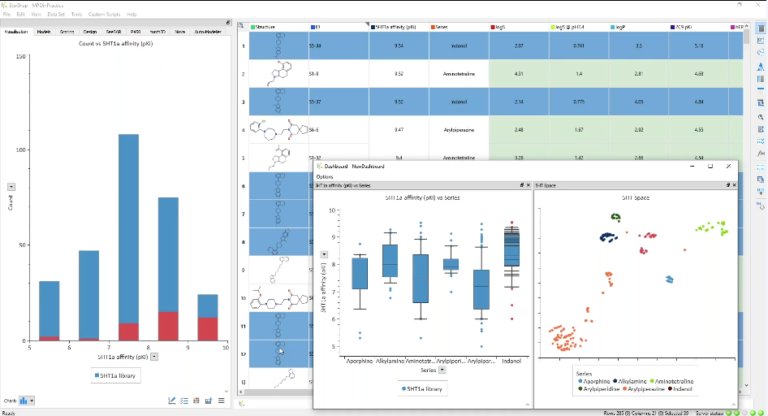
Accurate QSAR models lead to more efficient and cost-effective molecular discovery. Better predictions enable you to prioritise the optimal compounds…

The QuanSA method To define a ‘pocket field’, an initial alignment of all training molecules is constructed and function parameters…
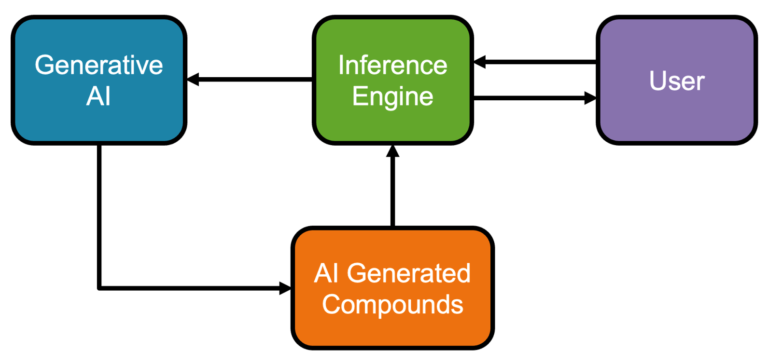
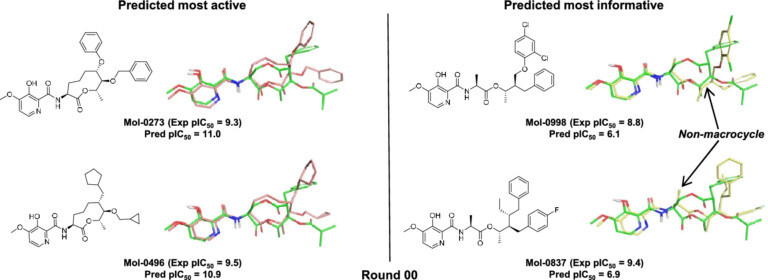
Pairing AI with human expertise We present a novel AI compound optimisation system, designed to include human oversight as a…
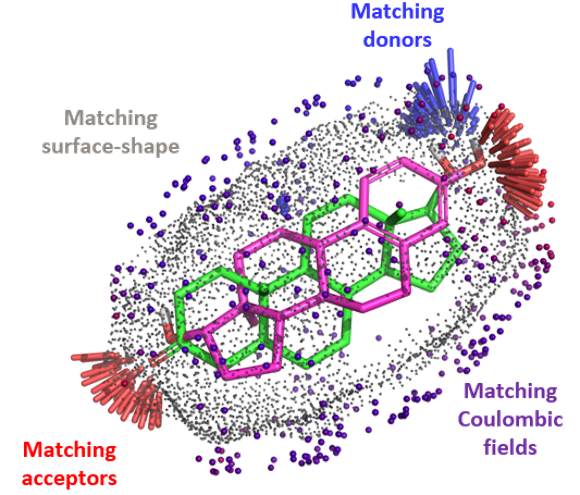
Introduction Using the integrated set of computational methods within the BioPharmics™ Platform, macrocycles can be effectively modelled for lead optimisation.…

To guide drug design, it’s important to understand the likely ADME and physicochemical properties of your compounds at an early…

Develop advanced MPO strategies and target the right compounds, faster.
We’re diving back into our favourite subject: multi-parameter optimisation.

Successful drugs require a delicate balance of many properties, such as potency, ADME and toxicity, to meet a project’s therapeutic objective. To make decisions about compound progression and assay selection, the available data must be assessed against project-specific criteria. However, the data on which we base our decisions often come from different sources and can vary in quality, so how can we use this information to make confident decisions? In addition, how can we be sure that the criteria we’re using are the most appropriate?

Scaffold replacement as part of an optimisation process that requires maintenance of potency, desirable biodistribution, metabolic stability, and considerations of synthesis at very large scale is a complex challenge.
Watch industry leaders from Novartis, Apollo Therapeutics and Eikon Therapeutics as they discuss their highs and lows, experience and advice,…

Interpreting metabolite-ID experiments; determining the right species for animal studies; providing optimisation suggestions for your medicinal chemistry colleagues to overcome…
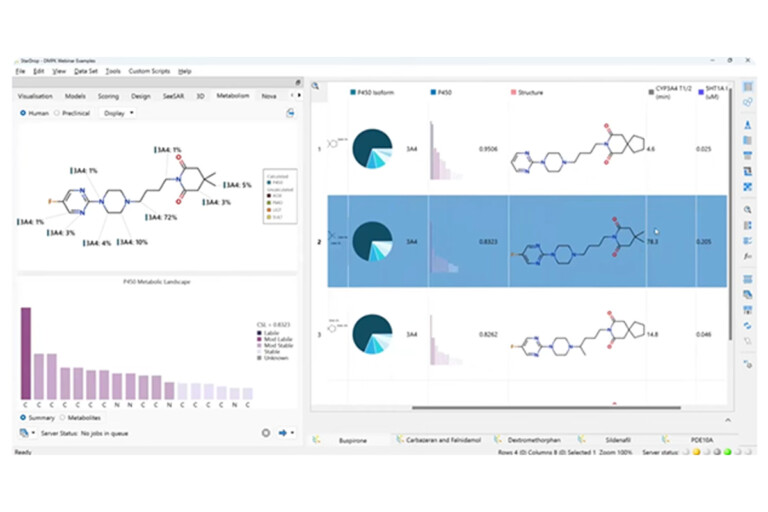
This peer-reviewed paper in Xenobiotica describes a new method to determine the most likely experimentally-observed routes of metabolism and metabolites based on our WhichP450™, regioselectivity and new WhichEnzyme™ model.
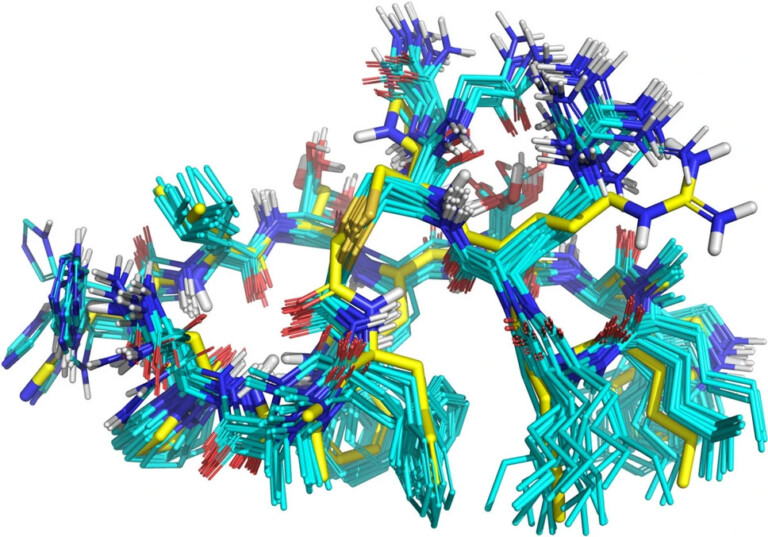
Systematic optimisation of large macrocyclic peptide ligands is a serious challenge. Here, we describe an approach for lead optimisation using the PD-1/PD-L1 system as a retrospective example of moving from initial lead compound to clinical candidate.
This paper describes a model to predict whether a particular site on a molecule will be metabolised by cytosolic sulfotransferase enzymes (SULTs).
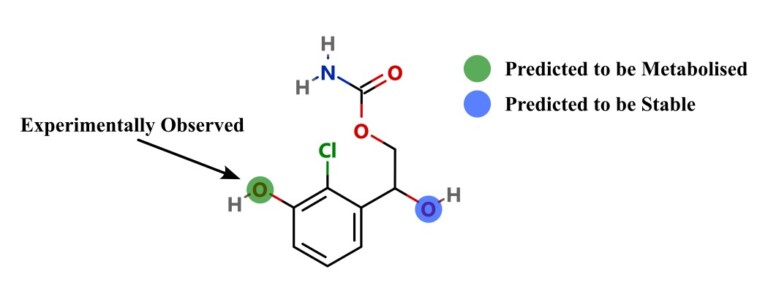
Introduction Predicting sites of metabolism (SoM) enable chemists to be more efficient in optimising the structure of new chemical entities…
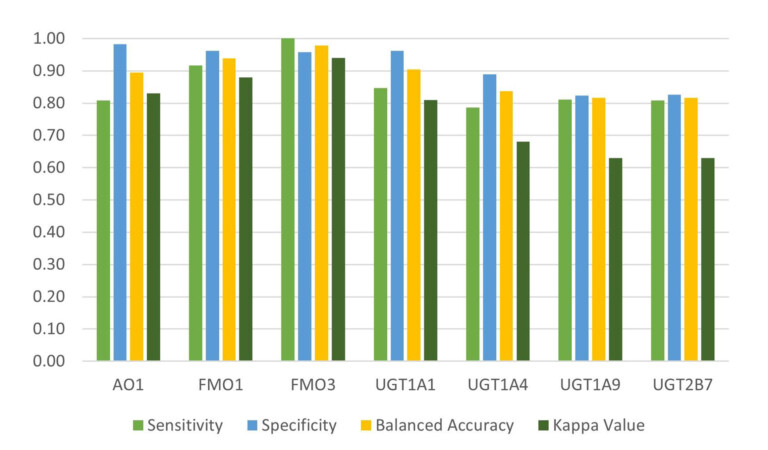
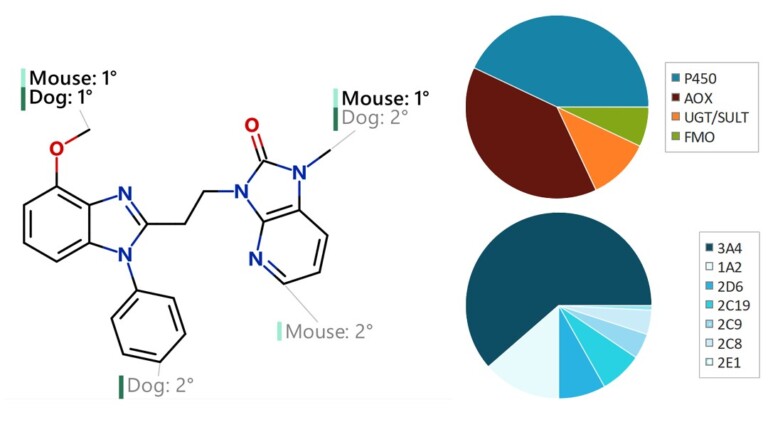
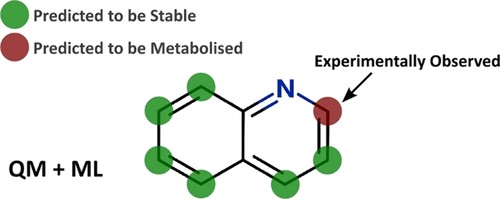
This paper describes the prediction of the regioselectivity of metabolism by AOs, FMOs and UGTs for humans and CYPs for three preclinical species.
In this webinar, we discuss Alchemite™, a novel deep learning approach, and its application to optimising kinase profiling programmes. The…

This webinar describes example applications of multi-parameter optimisation to find high-quality lead compounds.