Structure-based pose prediction: Non-cognate docking extended to macrocyclic ligands
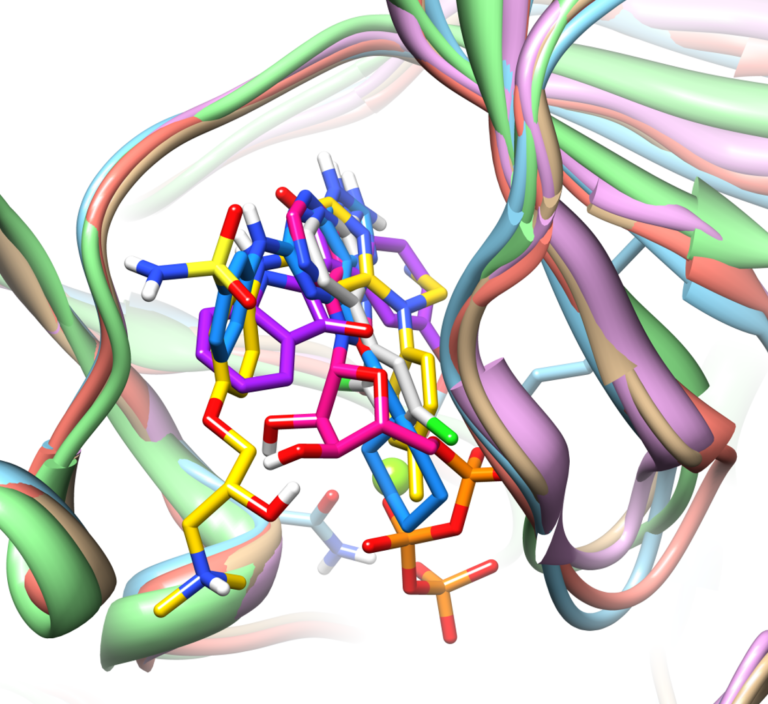
In this paper, we describe an extended benchmark for non-cognate docking of macrocyclic ligands, and the superior performance of Surflex™-Dock…
In this paper, we describe an extended benchmark for non-cognate docking of macrocyclic ligands, and the superior performance of Surflex™-Dock…
Interested in improving your binding mode predictions? Surflex™-Dock is a unique method for molecular docking, offering automatic pipelines for ensemble docking, applicable to both small molecules and large peptidic macrocycles alike.

Macrocycles are becoming increasingly popular in drug discovery, due to their vast potential against previously “undruggable” targets. But the size…
Scaffold replacement as part of an optimisation process that requires maintenance of potency, desirable biodistribution, metabolic stability, and considerations of synthesis at very large scale is a complex challenge.
Ann and Ajay discuss the science behind and applications of the eSim molecular similarity method, a ligand-based drug design approach which considers surface-shape, electrostatics, and directionally sensitive hydrogen-bonding when comparing two molecules.
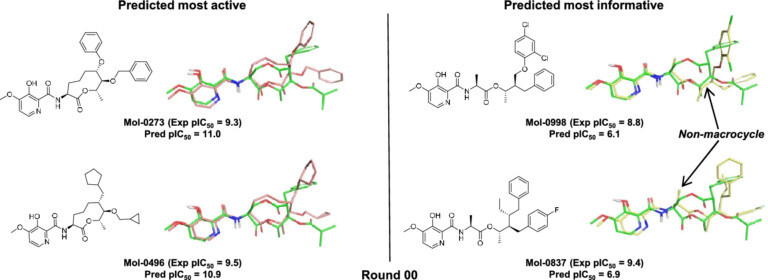
Systematic optimisation of large macrocyclic peptide ligands is a serious challenge. Here, we describe an approach for lead optimisation using the PD-1/PD-L1 system as a retrospective example of moving from initial lead compound to clinical candidate.
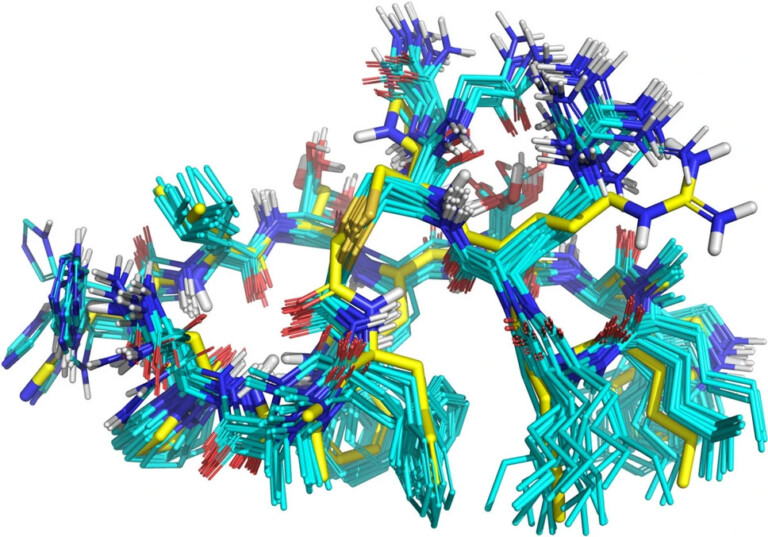
The solution structure of the minor conformer of rapamycin was investigated using a combination of NMR techniques and computational methods

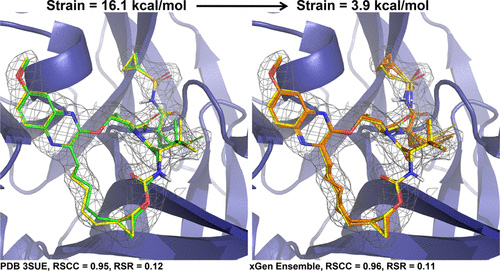
We show that the distribution of expected global strain energy values is dependent on molecular size in a superlinear manner. The distribution of strain energy follows a rectified normal distribution whose mean and variance are related to conformational complexity.
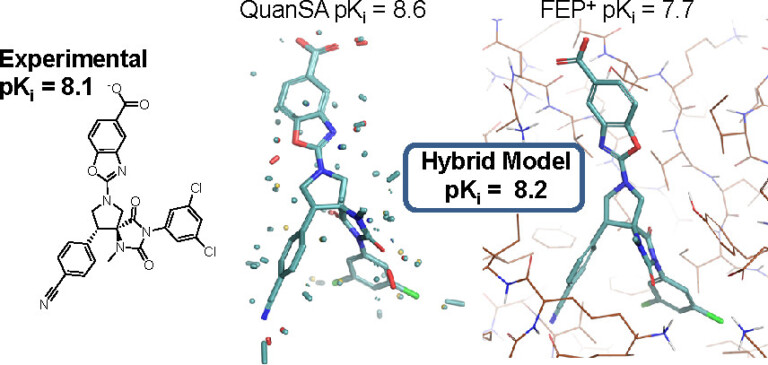
We present results on the extent to which physics-based simulation (exemplified by FEP+) and focused machine learning (exemplified by QuanSA) are complementary for ligand affinity prediction.
In this webinar, we demonstrate how Augmented Chemistry®, a unique deep learning method, can learn from higher throughput data together with limited panel data to provide high-quality imputations for sensory properties.
To better understand conformational propensities, global strain energies were estimated for 156 protein-macrocyclic peptide cocrystal structures.
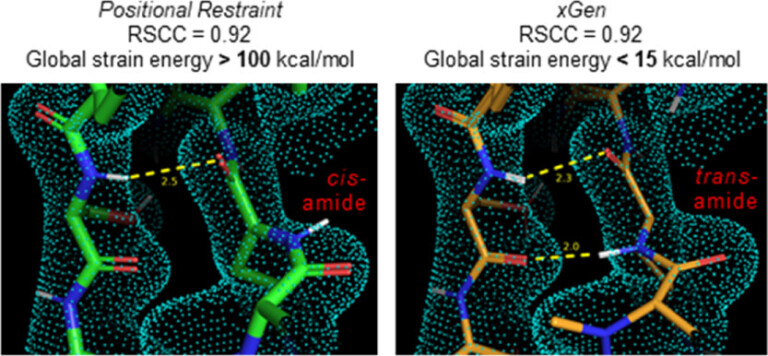
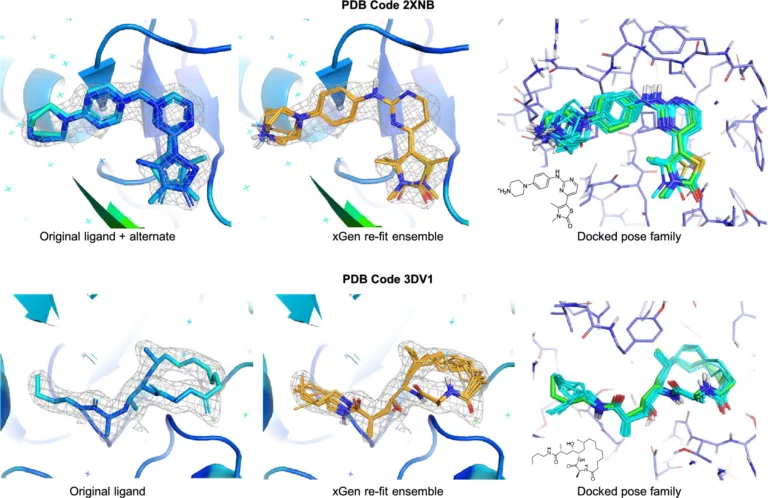
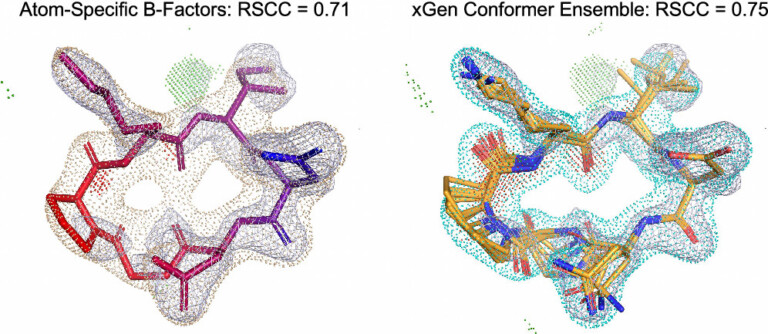
We report a new method for X-ray density ligand fitting and refinement that is suitable for a wide variety of small-molecule ligands, including macrocycles.
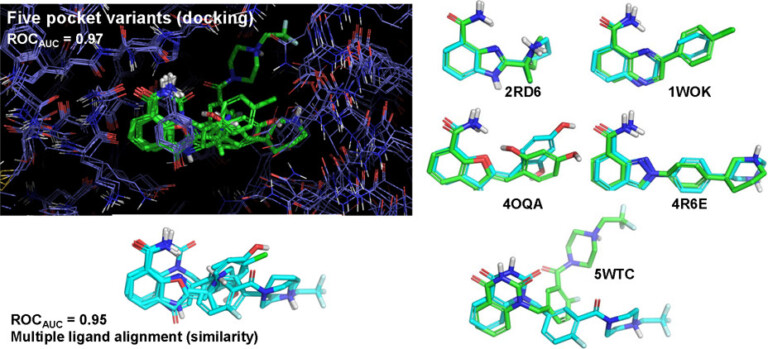
Using the DUD-E+ benchmark, we explore the impact of using a single protein pocket or ligand for virtual screening compared with using ensembles of alternative pockets, ligands, and sets thereof.
We introduce a new method for rapid computation of 3D molecular similarity that combines electrostatic field comparison with comparison of molecular surface-shape and directional hydrogen-bonding preferences (called “eSim”).
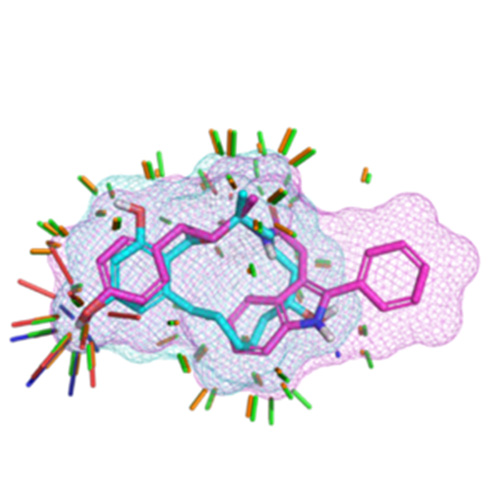
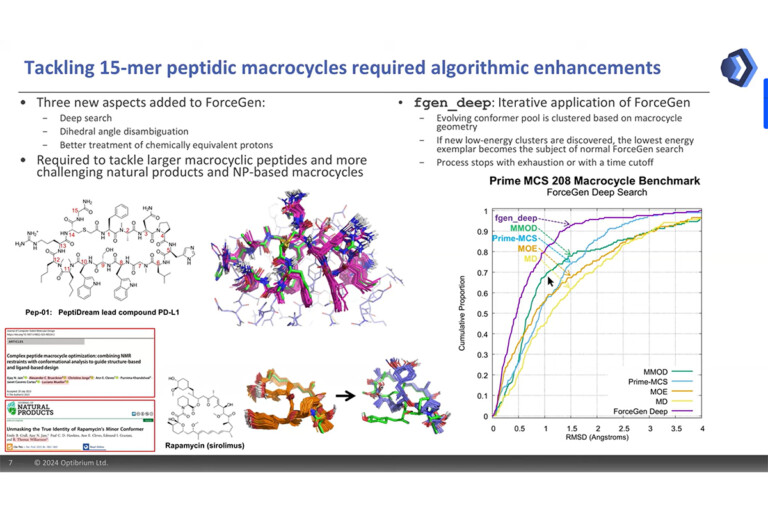
ForceGen is both faster and more accurate than the best of all tested methods on a very large, independently curated benchmark of 2859 PDB ligands. In this study, the primary results are on macrocycles, including results for 431 unique examples from four separate benchmarks.
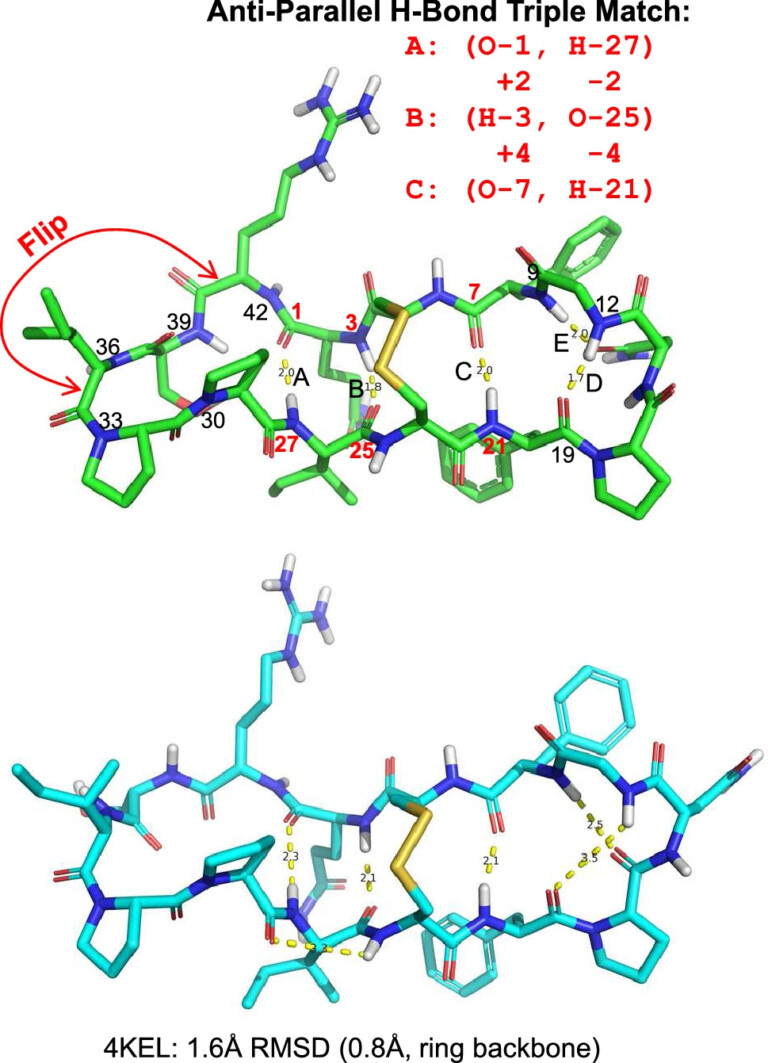
We introduce the QuanSA method for inducing physically meaningful field-based models of ligand binding pockets based on structure-activity data alone.

We introduce the ForceGen method for 3D structure generation and conformer elaboration of drug-like small molecules.
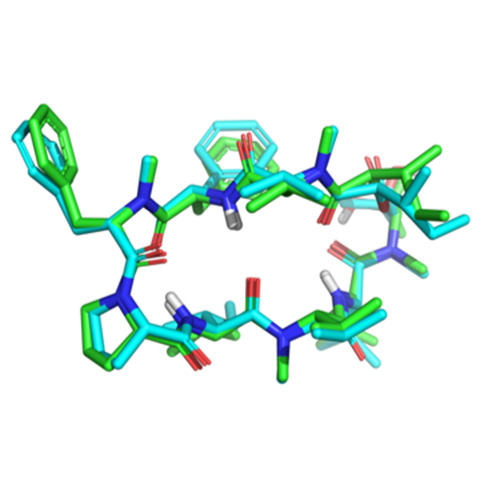
Surflex-QMOD integrates chemical structure and activity data to produce physically-realistic models for binding affinity prediction.
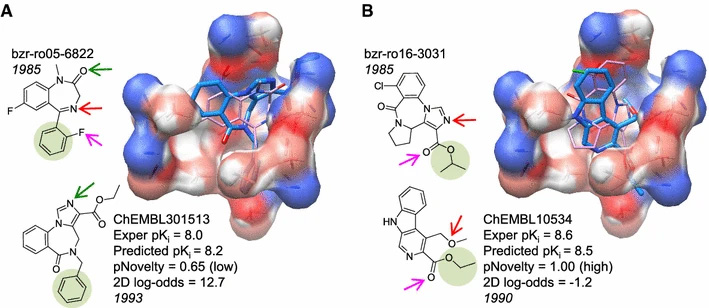
We present an approach that uses structural information known prior to a particular cutoff-date to make predictions on ligands whose bounds structures were determined later. The knowledge-guided docking protocol was tested on a set of ten protein targets using a total of 949 ligands.