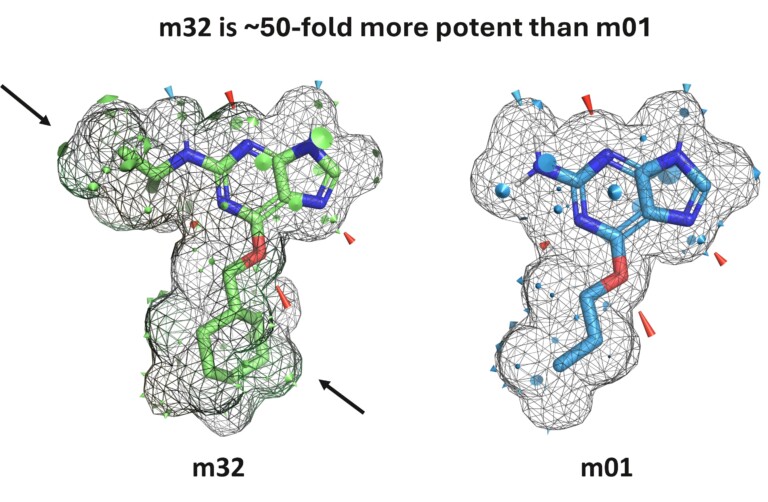
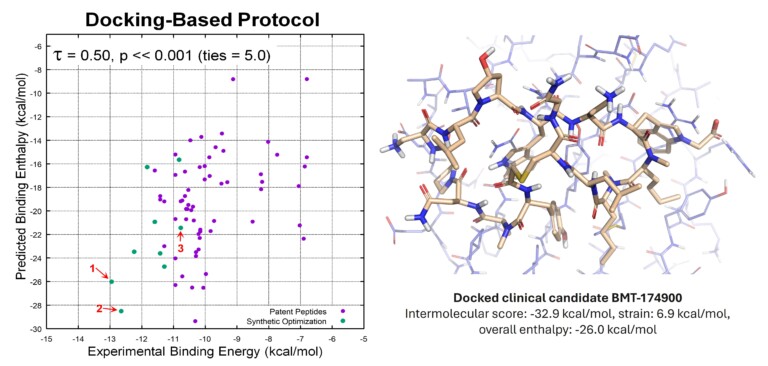
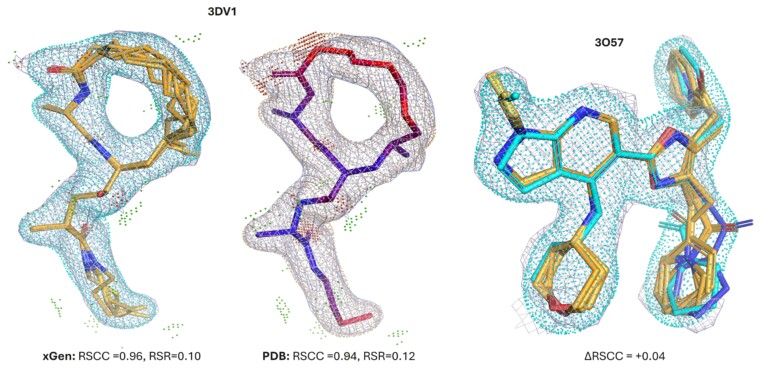
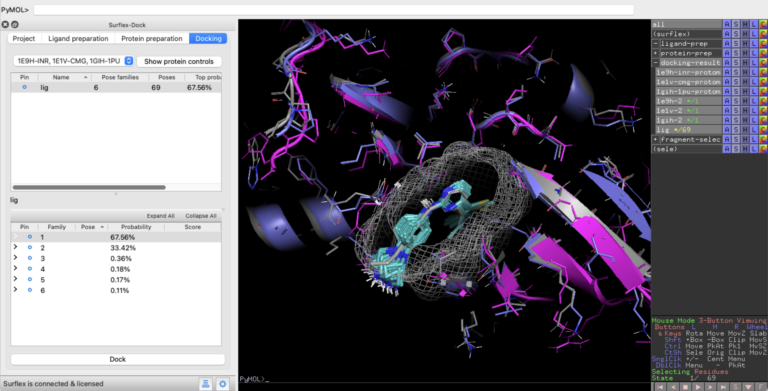
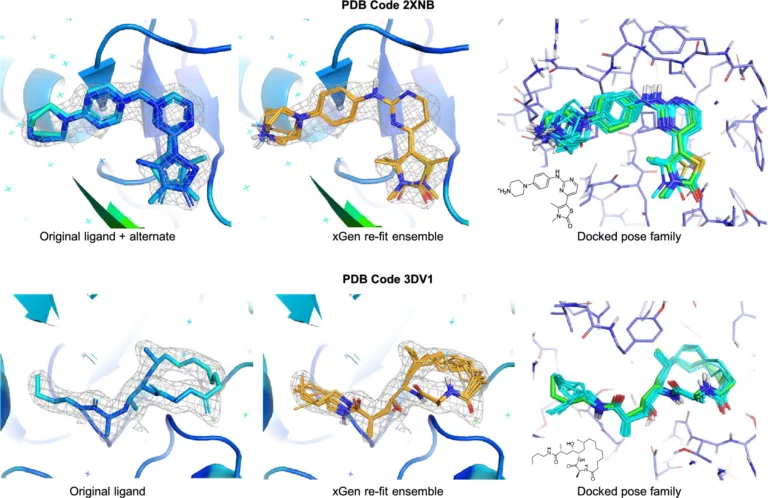
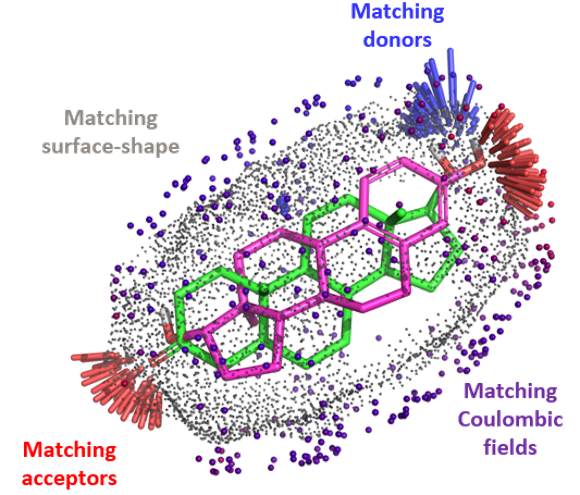
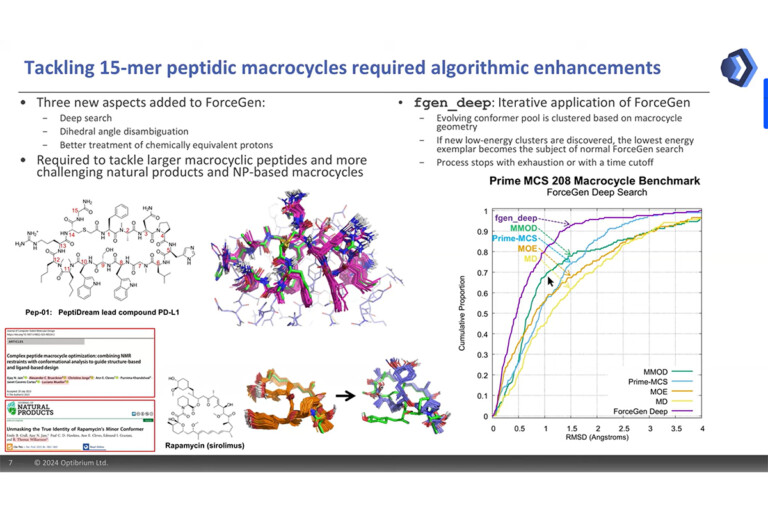
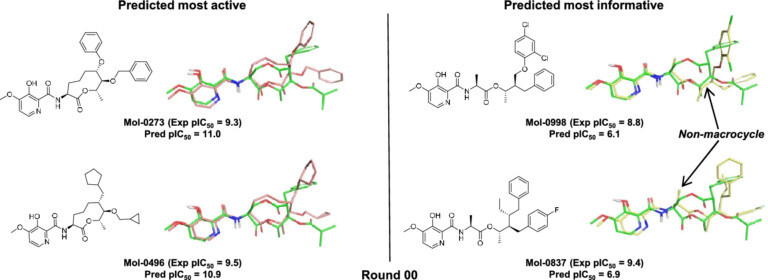
Optibrium Introduces Graphical Interface for QuanSA to Enhance Ligand-Based Affinity Predictions
New user interface provides visual insights to guide compound optimisation CAMBRIDGE, UK, 24 March 2026 – Optibrium, a leading developer of software and AI solutions for molecular design, today…