Why should medicinal chemists use software?
What exactly is a medicinal chemist’s role today? Firstly, let’s define a medicinal chemist’s role – to design and synthesise…

What exactly is a medicinal chemist’s role today? Firstly, let’s define a medicinal chemist’s role – to design and synthesise…
It feels like only yesterday that we announced the arrival of StarDrop 8. And here we are again. Though this time, that’s rather the point. Discovery teams need to iterate quickly, moving from hypothesis…

Download the guide and learn what generative chemistry is, where it fits in the discovery workflow, and best practices to avoid common pitfalls.

Why traditional ligand-based QSAR methods have fallen short You might be sceptical about ligand-based QSAR approaches; many researchers are. We discussed why there has…

Imagine you’re trying to find the correct key to unlock a treasure box, but there are billions of keys to…

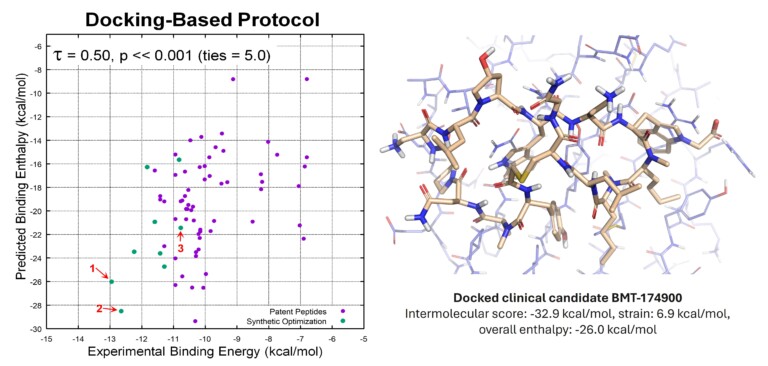
From X-ray refinement to lead optimisation Conventional ligand-fitting and refinement methods in X-ray electron density maps often yield models with…
The collaboration challenge: Where generic tools fall short Several collaboration platforms have been designed to support molecule design and optimisation.…

How number of users affect drug discovery software costs The number of people who need access to the platform is…

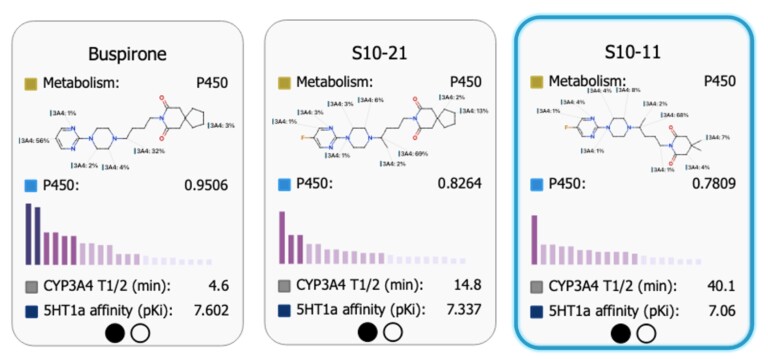
Good statistics don’t guarantee good applicability. Performance metrics will tell you how well a QSAR model predicts known data, but they don’t tell you whether it will add practical value…
