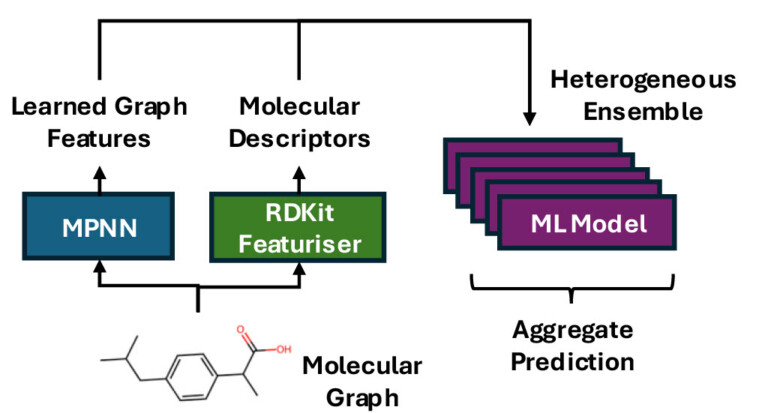
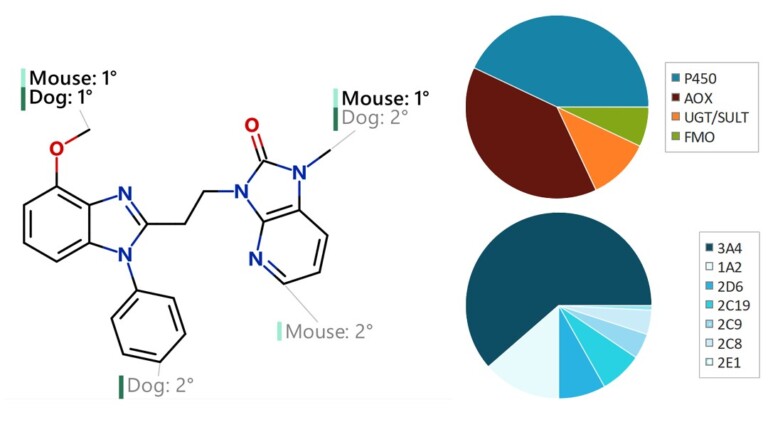
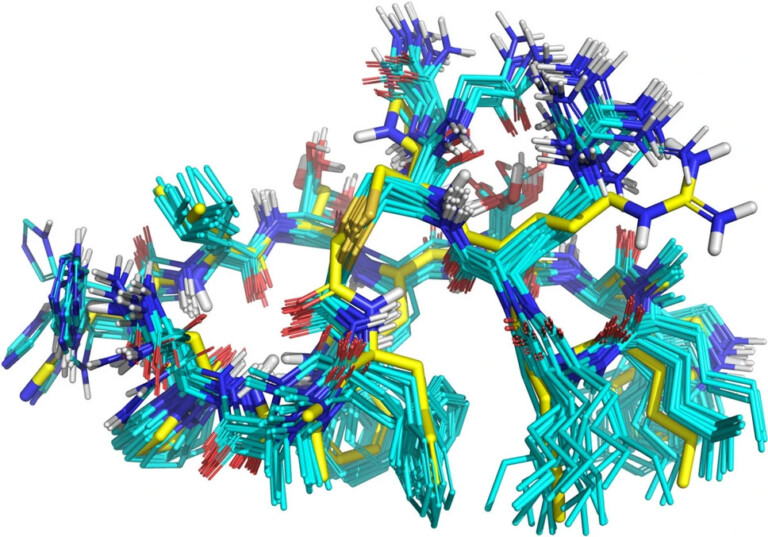
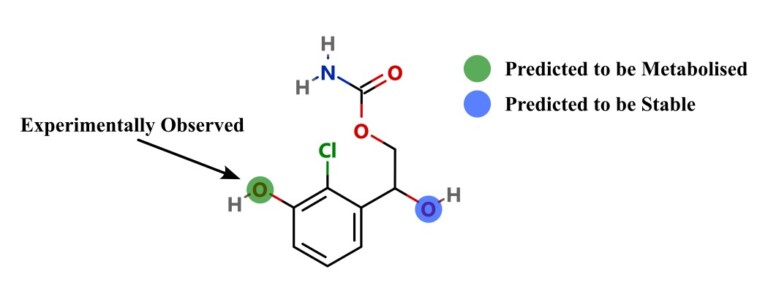
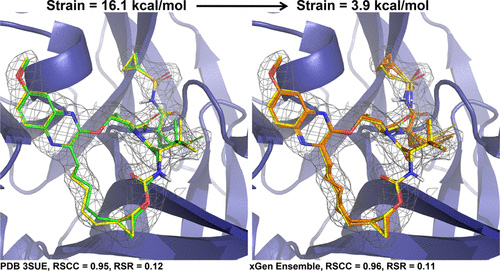
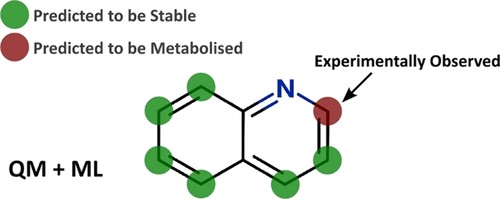
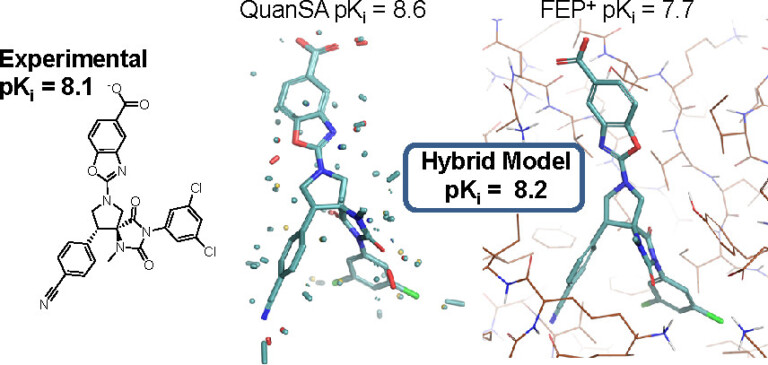
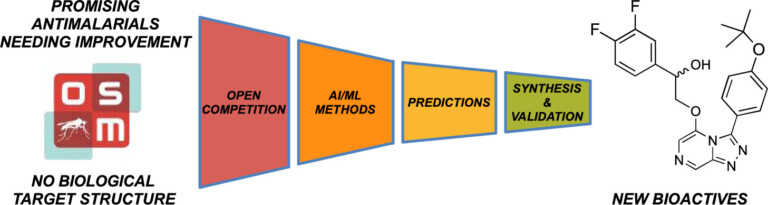
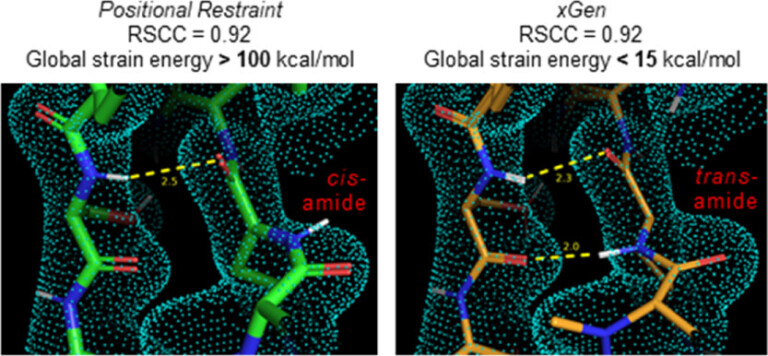
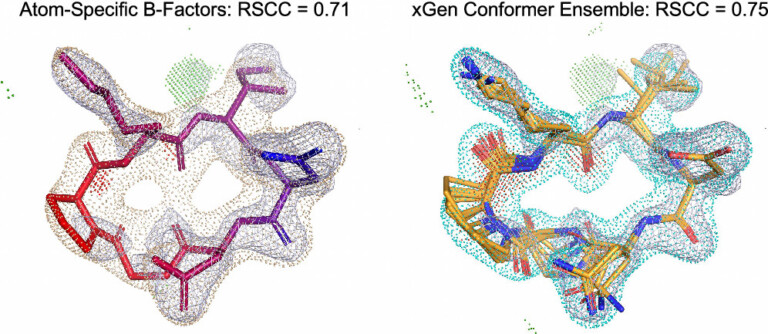
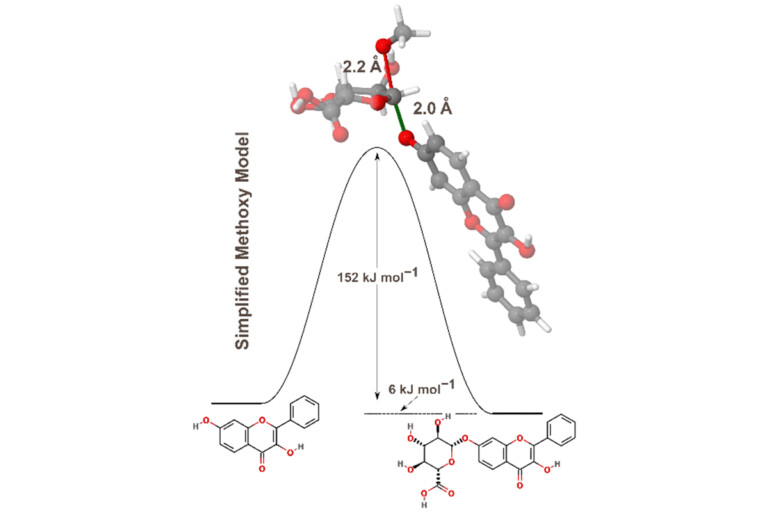
Improving predictions of molecular properties with graph featurisation and heterogeneous ensemble models
Abstract We explore a “best-of-both” approach to modelling molecular properties by combining learned molecular descriptors from a graph neural network…