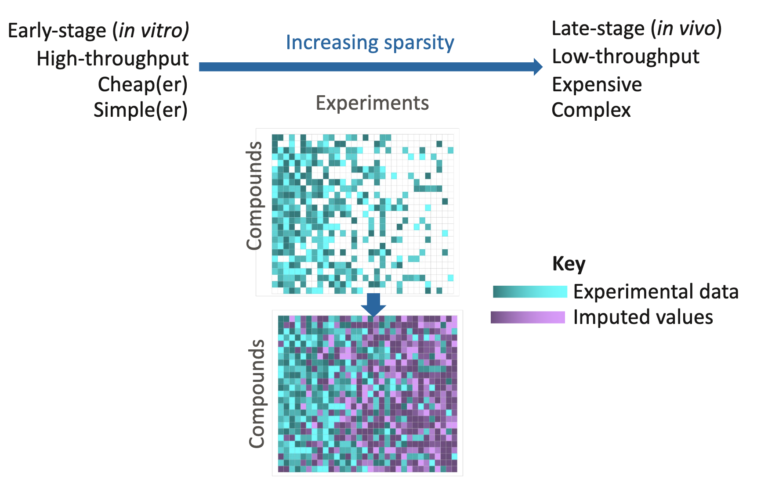
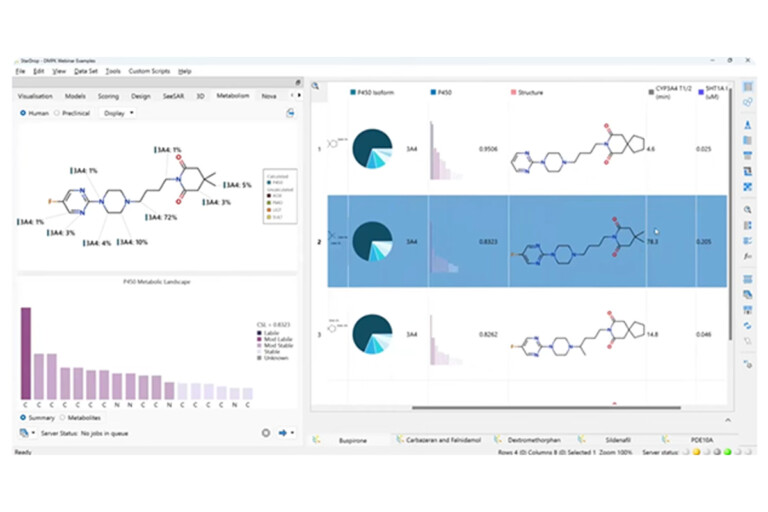
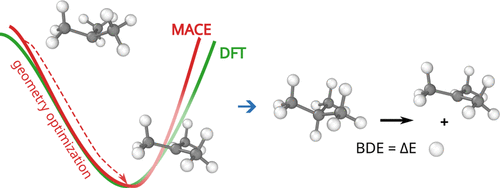
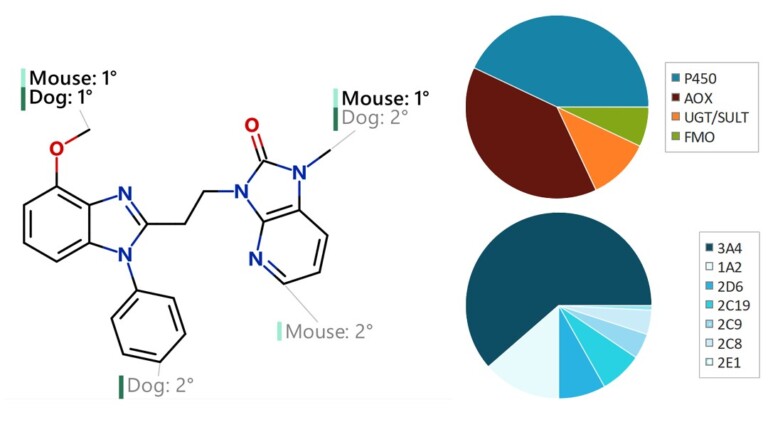
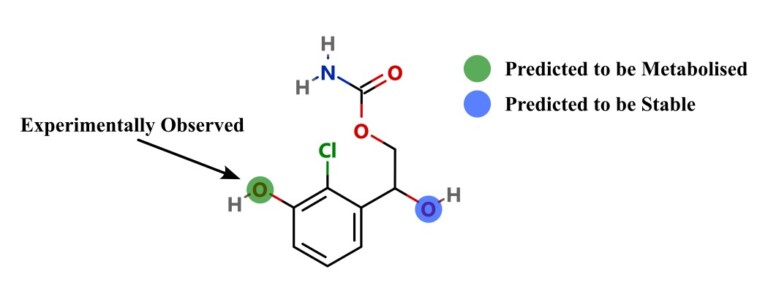
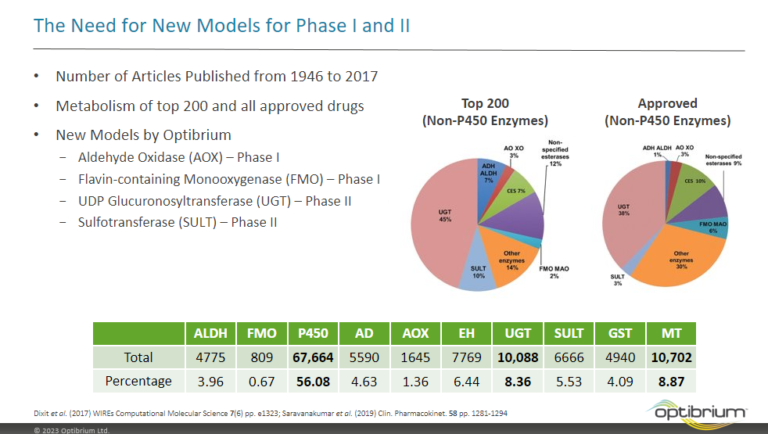
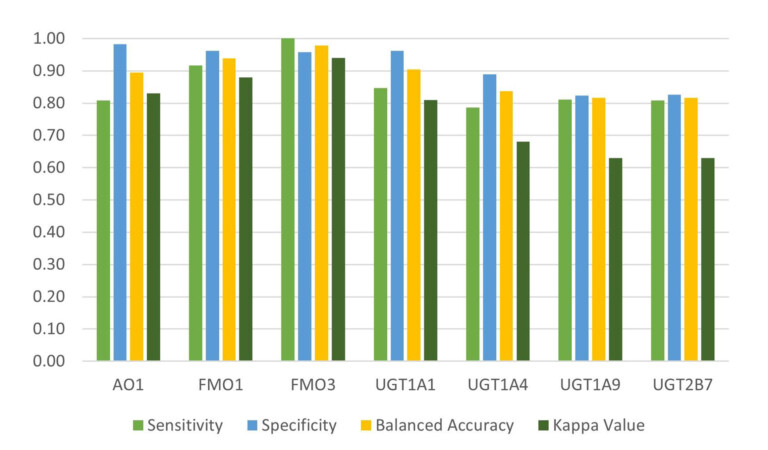
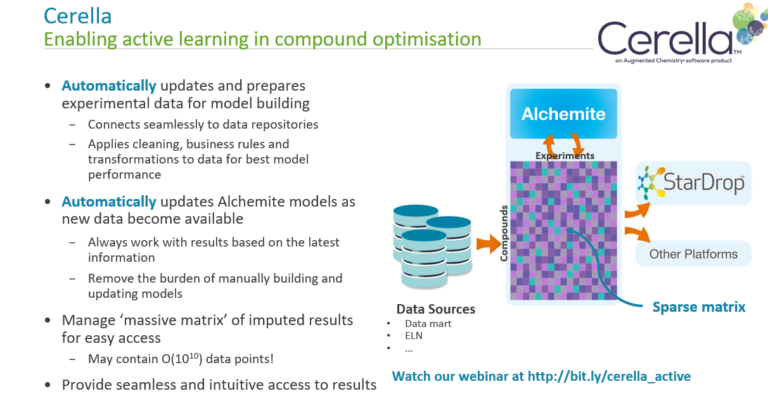
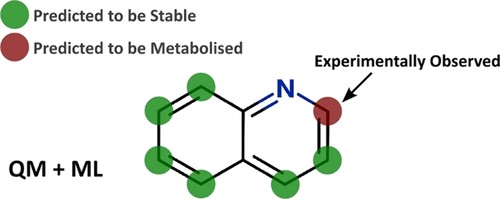
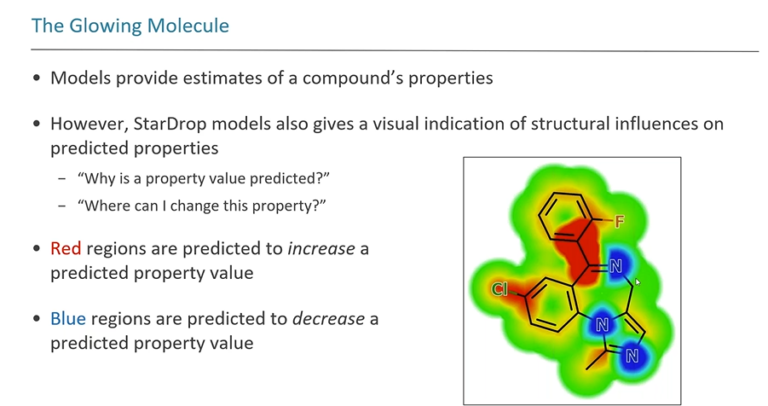
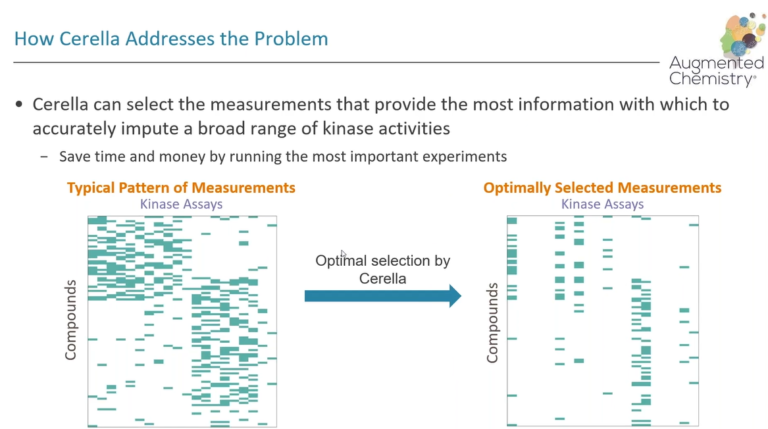
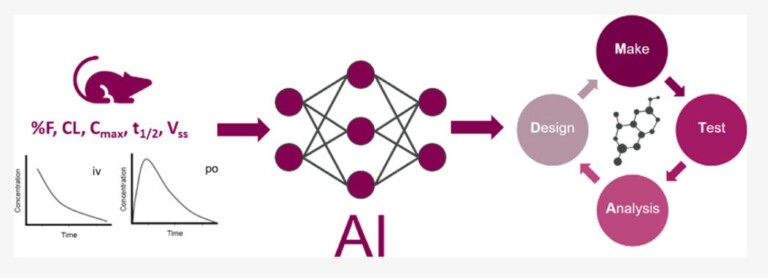
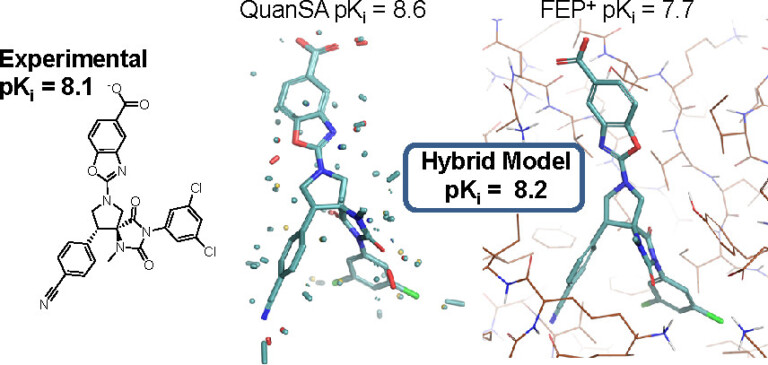
In this webinar, we explore the highlights of collaborative project results that demonstrate how every phase of the drug discovery process can be radically improved by applying proven AI technology. Providing scientists with insights on which to base decisions can identify valuable new opportunities and reduce the time and cost of AI drug discovery cycles.
We review case studies from collaborations with Constellation Pharmaceuticals, AstraZeneca, Genentech, the University of Dundee and Takeda Pharmaceuticals to validate the impact of applying AI to experimental data and illustrate dramatic improvements to their project outcomes.
Join Samar Mahmoud and Matt Segall for this fascinating deep dive into the revolution that AI is bringing to the challenges of sparse and noisy drug discovery data.