Dramatically cut project timelines and costs with BioPharmics™, proven to reduce synthesis requirements by up to 90% [1]
Our technology delivers unparalleled speed and accuracy, matching the high standards of small molecule modelling and orders of magnitude faster than any competing solution[2,3]. The result? BioPharmics is the only platform that enables you to routinely incorporate 3D macrocycle modelling into your hit-to-candidate workflows.
By slashing synthesis and testing requirements, BioPharmics’ industry-leading 3D modelling technology accelerates project timelines, lowers costs and increases success rates in macrocycle discovery.
With over 40 publications to date, and trusted by top 10 pharma companies to solve complex macrocycle challenges.
BioPharmics: Finally, macrocycle modelling that actually works
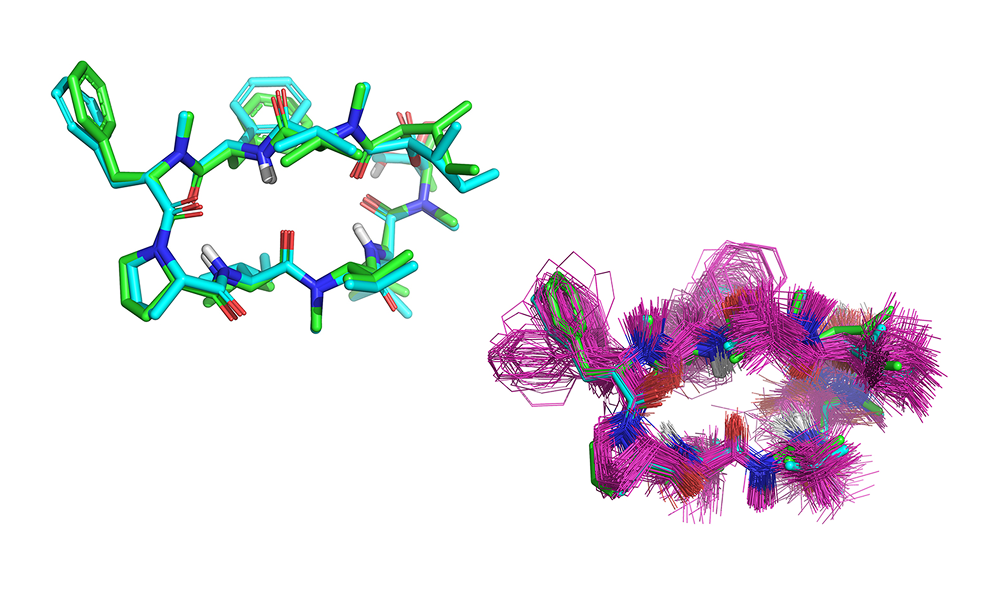
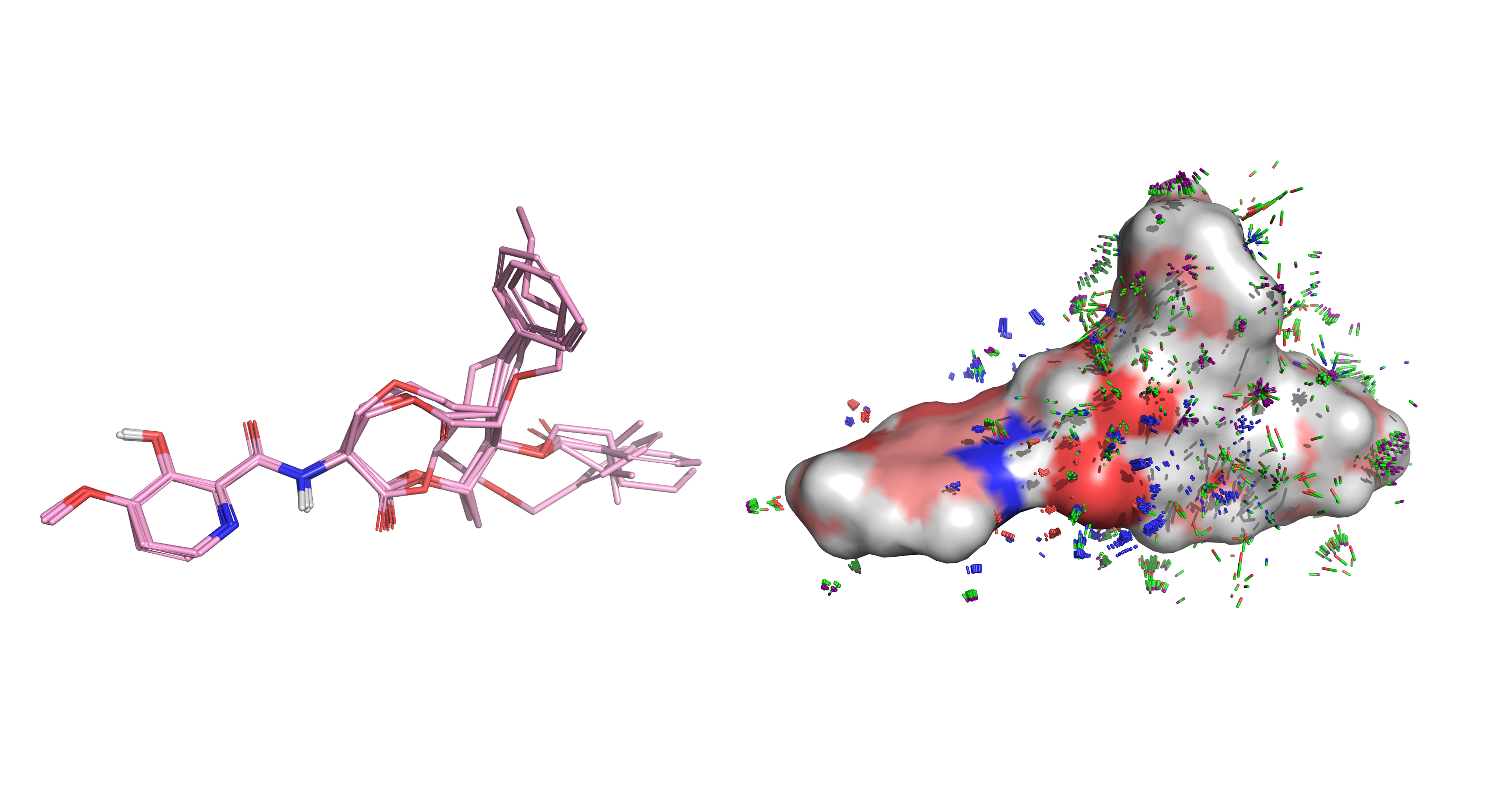
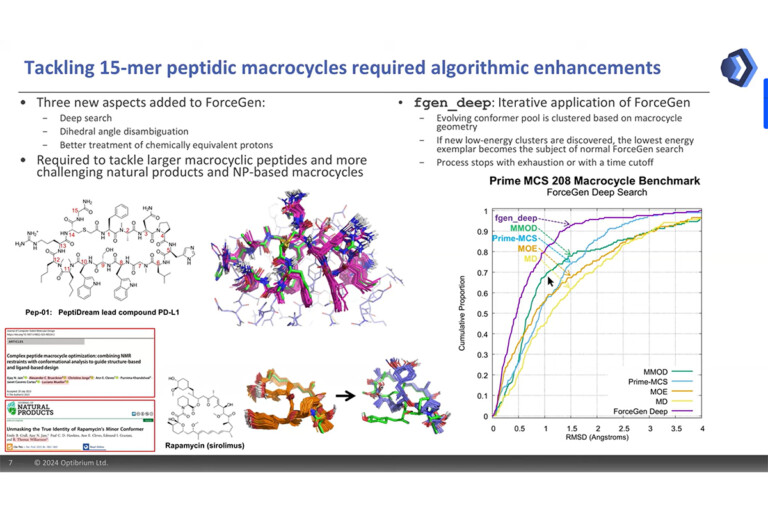
BioPharmics’ advanced conformational sampling algorithms capture macrocycle flexibility with a speed and rigour that other software simply cannot provide. By generating low-energy conformational ensembles, with the option to incorporate experimental biophysical data from NMR, X-ray, or cryo-EM,and applying state-of-the-art structure- and ligand-based methods, BioPharmics delivers unparalleled accuracy in identifying active peptide macrocycles and correctly predicting their bindingposes.
BioPharmics’ advanced algorithms excel in comprehensive, high-quality conformational sampling, critical for successful macrocycle modelling.
BioPharmics: Your comprehensive macrocycle modelling suite
Our industry-leading 3D ligand and structure-based design software is fully inclusive of five specialised methodologies:
ForceGen™: Generate complete conformational ensembles in minutes
Our patented ForceGen method uses physically intuitive molecular movements to explore the conformational space of molecules, without the need to rely on precalculated molecular templates or torsional libraries. This means that it can rigorously and efficiently explore the conformational space of even large, flexible macrocycles. It generates low-energy conformational ensembles with a high likelihood of containing close matches to experimental conformations. It works with simple SMILES input and supports the integration of experimental biophysical data to effectively identify solution-state and bioactive conformations [1,4].
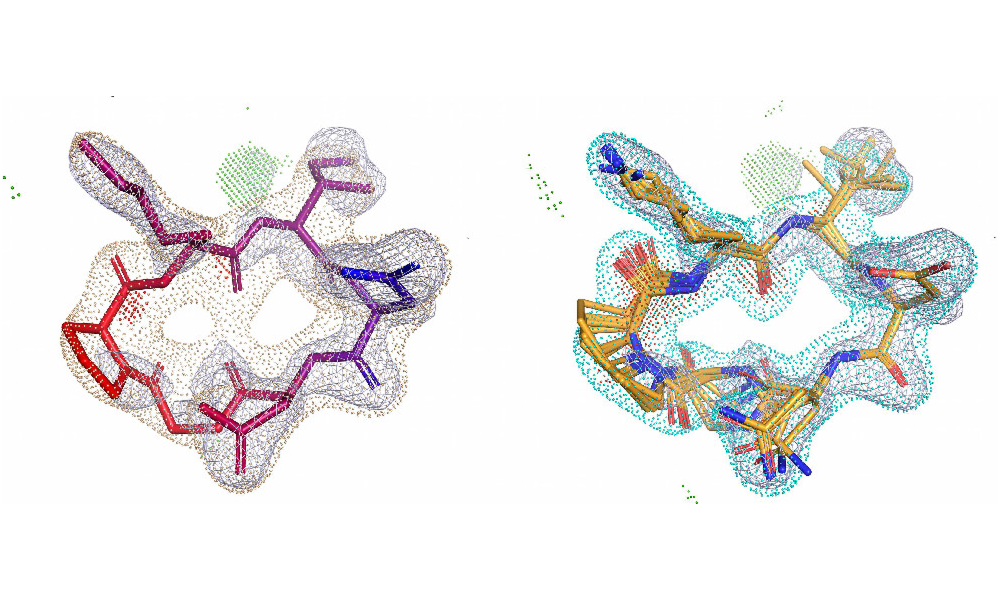
xGen™: Refine conformational ensembles with x-ray electron density maps
xGen is our real-space ligand refinement method, which seeks low-energy solutions to fitting ligand X-ray density [4]. It overcomes the limitation of other ligand density fitting methods to produce more accurate, lower-strain models of how ligands bind in the active site, which is critical when working with macrocycles [5]. xGen is accessible to non-crystallographers and within crystallographic workflows.
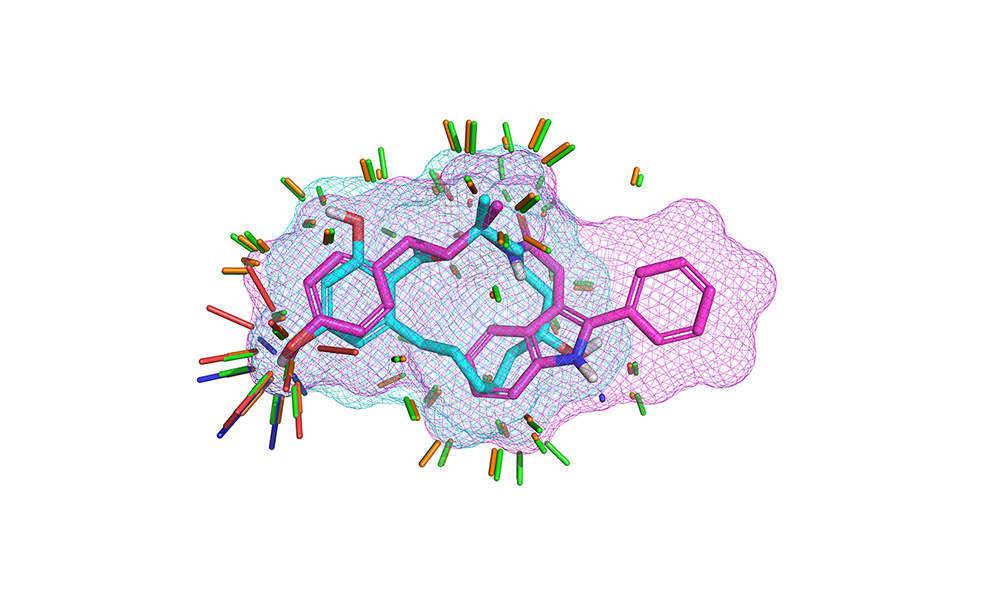
eSim™: High performance 3D virtual screening
Want to find new active compounds but don’t have a target structure? Perform 3D structural alignment with eSim that leverages ForceGen’s conformational power. eSim analyses molecular shape, charge distribution, and binding preferences to screen millions of compounds with superior hit rates.
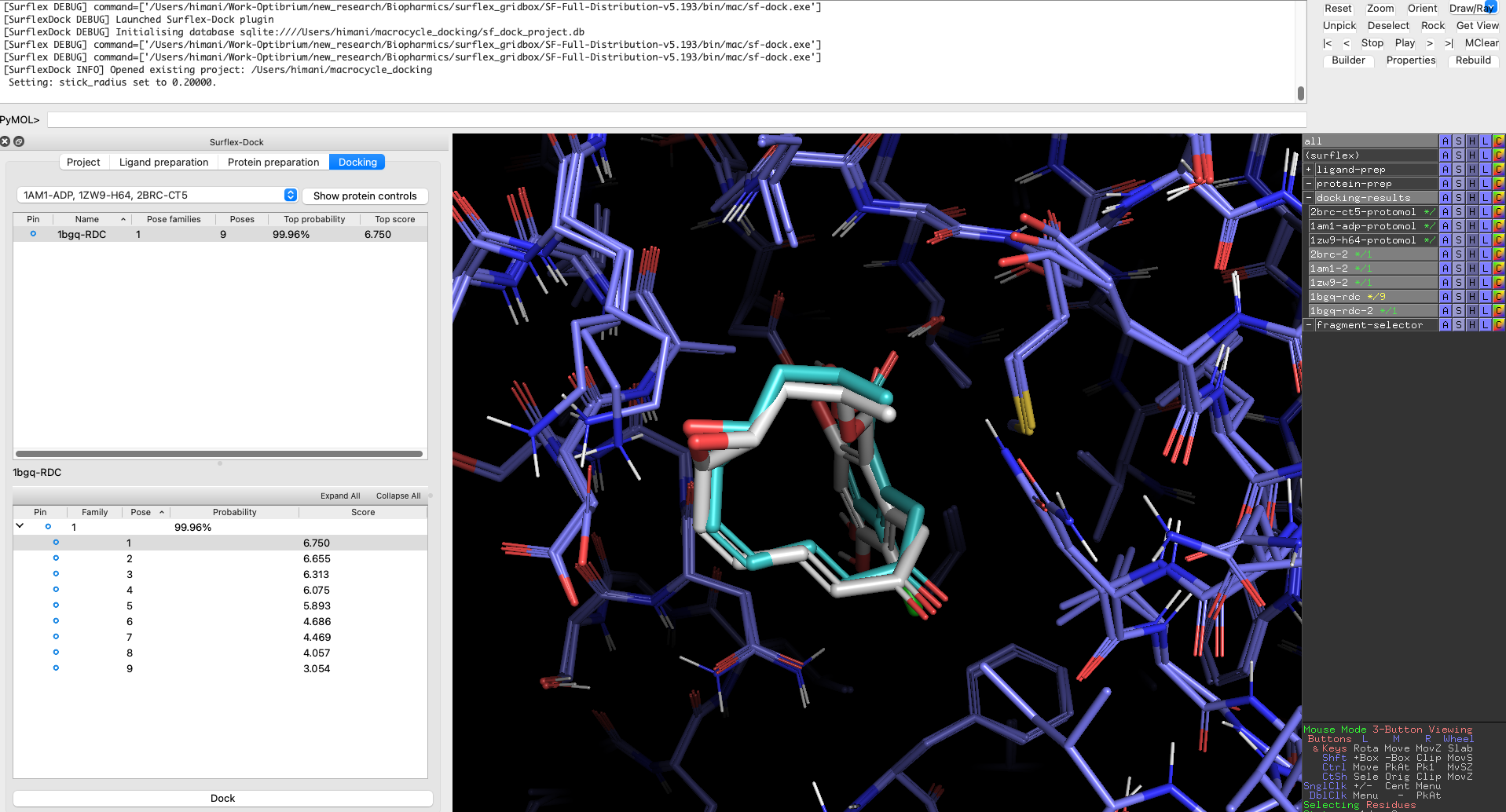
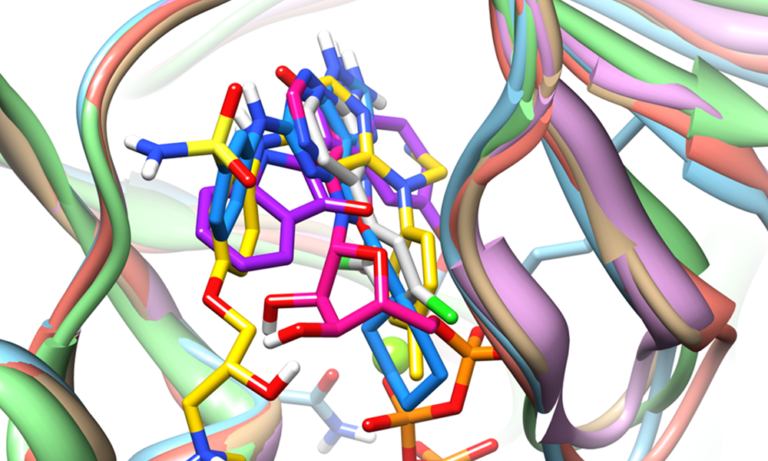
Our advanced, fully-automated docking method excels at predicting macrocycle-target interactions [3]. Surflex-Dock combines efficient conformational sampling for flexible ligands with optimised scoring functions, and can use prior knowledge of bound ligands to generate more accurate predictions of bound poses.
Combined with a model of strain energy, this effectively ranks compounds by affinity, enabling you to prioritise macrocycles for synthesis and testing, thereby saving time and synthetic effort [1].
QuanSA™: Accurate ligand-based binding affinity prediction
Our unique 3D ligand-based method for predicting affinity has comparable accuracy to FEP+, is 1000x faster and has a broader domain of applicability, enabling you to make accurate predictions for new scaffolds and series [8,9]. All of this without the need for a protein target structure! It does this by constructing surface field-based models of ligand binding pockets and using multiple instance machine learning to account for potential variations in compound poses. And, if you do perform FEP simulations, QuanSA results are complementary at a fraction of the computational cost – the combination of QuanSA and FEP is more accurate than either individually.
Proven science, peer-reviewed results
BioPharmics’ modelling capabilities are backed by rigorous scientific validation and published research. We provide a more comprehensive list of references below, but here are a few highlights:
Structure-based pose prediction: Non-cognate docking extended to macrocyclic ligands
This paper demonstrates that predicting the binding poses of macrocyclic ligands in protein pockets (cross-docking) is as successful as for non-macrocyclic compounds, even when the protein structures are “macrocycle-naive”. Using the ForceGen and Surflex-Dock methods, approximately 80% of poses were correctly predicted within the top two pose families, significantly outperforming other tools. This demonstrates a significant advance for computational macrocycle design.
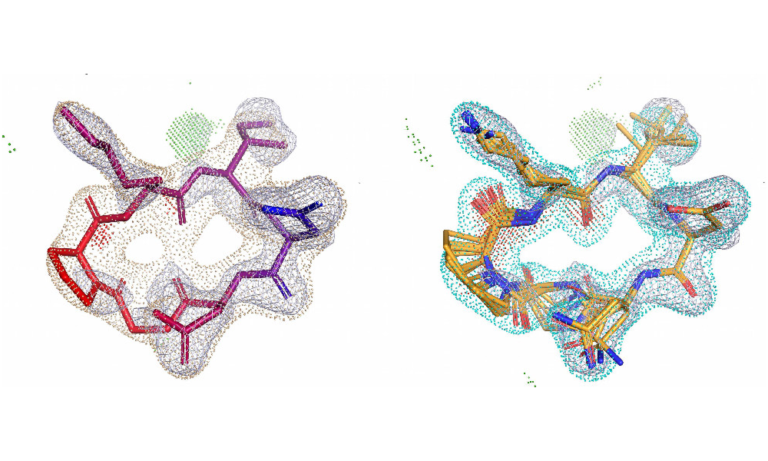
Complex peptide macrocycle optimisation: combining NMR restraints with conformational analysis to guide structure-based and ligand-based design
Together with BMS, this paper details a multi-disciplinary approach for optimising large macrocyclic peptides, combining NMR-derived conformational restraints with computational analysis. It enables accurate prediction of bound ligand poses and conformational strain, revealing that subtle structural changes significantly impact strain, which critically affects binding. This combination enableseffective prioritisation of lead compound analogues, saving synthetic effort by 90%.
A Distributional Model of Bound Ligand Conformational Strain: From Small Molecules up to Large Peptidic Macrocycles
In collaboration with Merck and BMS, this study uses xGen real-space refinement to reveal that bound ligand conformational strain energies are significantly lower and more plausible than prior estimates from deposited PDB structures. It introduces a size-dependent distributional model for strain, which is superlinear, offering improved guidance for conformational search and molecular design across diverse ligands.
Introduction Using the integrated set of computational methods within the BioPharmics™ Platform, macrocycles can be effectively modelled for lead optimisation.…
Macrocycles are becoming increasingly popular in drug discovery, due to their vast potential against previously “undruggable” targets. But the size…
Speak to our team to find out how you can reduce synthesis requirements by up to 90%
BioPharmics is proven to cut synthesis requirements by up to 90% and deliver accurate pose prediction for even the most complex macrocycles. Speak with our team to learn how our technology can support your hit-to-candidate workflows and dramatically accelerate timelines.
References
[1] Jain, A.N., Brueckner, A.C., et al. (2023) J. Comput.-Aided Mol. Des., 37, 519-535 [2] Jain, A.N., Cleves, A.E., et al. (2019) J. Comput.-Aided Mol. Des. , 33, 531-558 [3] Cleves, A.E., Tandon, H. and Jain, A.N. (2024), J. Comput.-Aided Mol. Des., 38, 33 [4] Jain, A.N., Cleves, A.E., et al. (2020) J. Med. Chem., 63,10509−10528 [5] Jain, A.N., Brueckner, A.C., et al. (2023) J. Med. Chem., 66,1955–1971 [6] Cleves, A.E., Johnson, et al. (2019) J. Comput.-Aided Mol. Des., 33, 865-886 [7] Cleves, A.E. and Jain, A.N. (2020) J. Chem. Inf. Model., 60, 4296–4310 [8] Cleves, A.E., Johnson, S.R., and Jain, A.N. (2021) J. Chem. Inf. Model., 61, 5948-5966 [9] Cleves, A.E., Jain, A.N., et al. (2024) J. Comput.-Aided Mol. Des., 38, 19