Why does conformational flexibility matter in drug design?
What are conformational ensembles? A conformational ensemble is a collection of the different 3D shapes a molecule can adopt in…

How much does 3D molecular modelling software cost?
Introduction 3D molecular modelling plays a vital role in modern drug discovery, offering powerful applications to streamline research, reduce costs,…

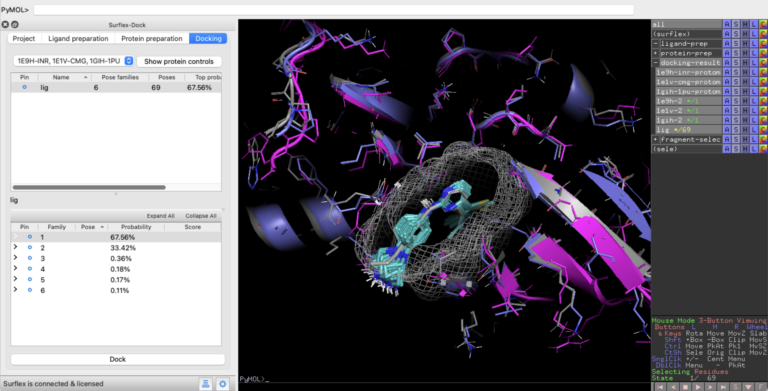
Optibrium Widens Access to Industry-Leading Docking Method
New PyMOL interface extends access to Optibrium’s structure-based design method Surflex™-Dock CAMBRIDGE, UK, 04 February 2025 – Optibrium, a leading…
How can I model large molecules like macrocycles?
What comprises large molecules? When we talk about “large molecules,” we often think of biologics like monoclonal antibodies, proteins, and…

Optibrium demonstrates superior molecular docking method for small molecules and macrocycles
CAMBRIDGE, UK, 22 October 2024 – Optibrium, a leading developer of software and AI solutions for molecular design today announced…
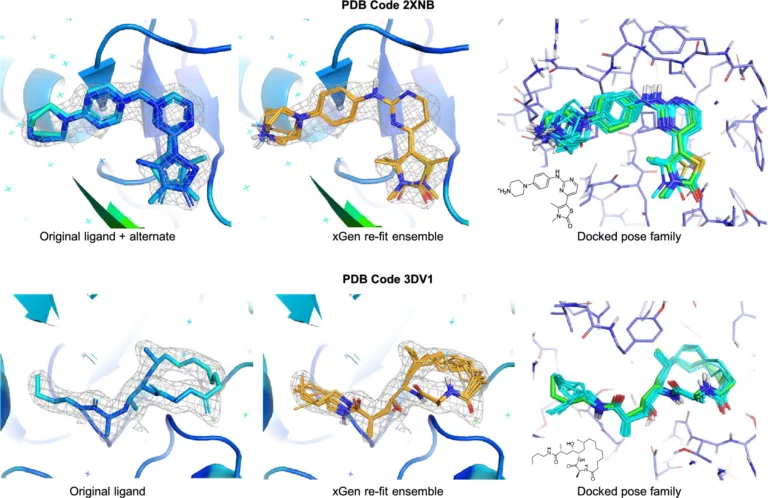
Macrocyclic peptide optimisation: Integrating computational approaches with biophysical data
Introduction Using the integrated set of computational methods within the BioPharmics™ Platform, macrocycles can be effectively modelled for lead optimisation.…

Structure-based pose prediction: Non-cognate docking extended to macrocyclic ligands
In this paper, we describe an extended benchmark for non-cognate docking of macrocyclic ligands, and the superior performance of Surflex™-Dock…
Molecular docking: Extrapolating to new scaffolds with Surflex™-Dock
Interested in improving your binding mode predictions? Surflex™-Dock is a unique method for molecular docking, offering automatic pipelines for ensemble docking, applicable to both small molecules and large peptidic macrocycles alike.

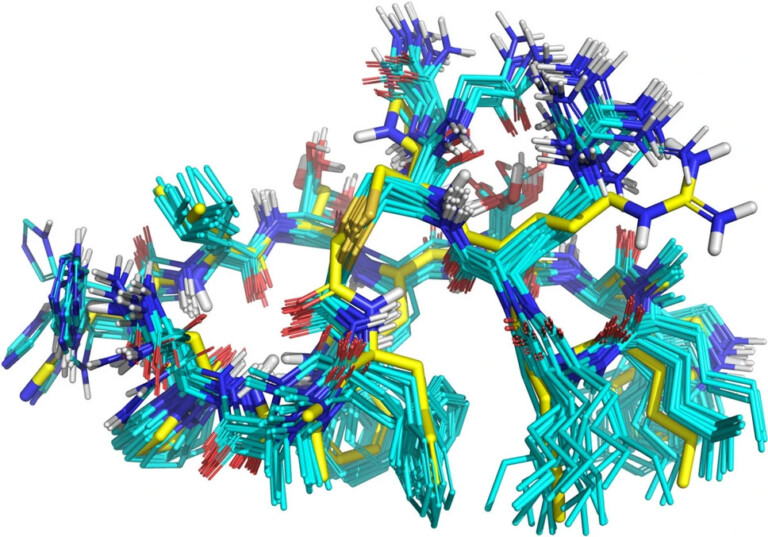
Complex peptide macrocycle optimisation: combining NMR restraints with conformational analysis to guide structure-based and ligand-based design
Systematic optimisation of large macrocyclic peptide ligands is a serious challenge. Here, we describe an approach for lead optimisation using the PD-1/PD-L1 system as a retrospective example of moving from initial lead compound to clinical candidate.
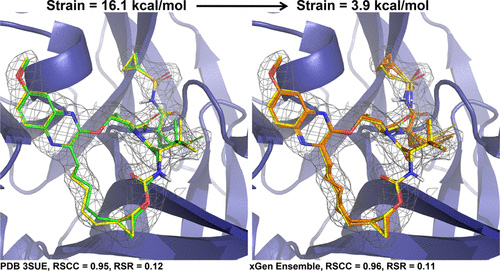
A distributional model of bound ligand conformational strain: from small molecules to large peptidic macrocycles
We show that the distribution of expected global strain energy values is dependent on molecular size in a superlinear manner. The distribution of strain energy follows a rectified normal distribution whose mean and variance are related to conformational complexity.
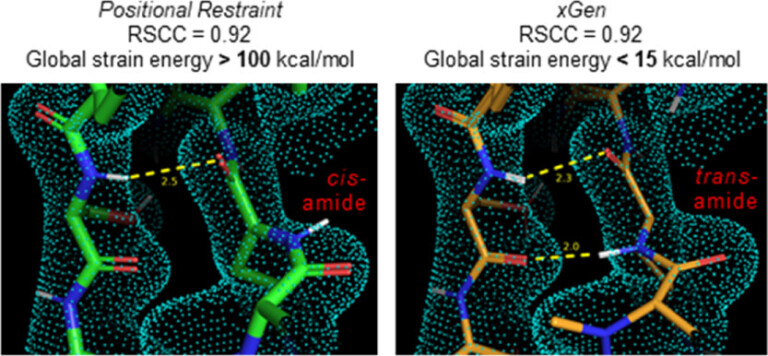
Conformational strain of macrocyclic peptides in ligand–receptor complexes based on advanced refinement of bound-state conformers
To better understand conformational propensities, global strain energies were estimated for 156 protein-macrocyclic peptide cocrystal structures.
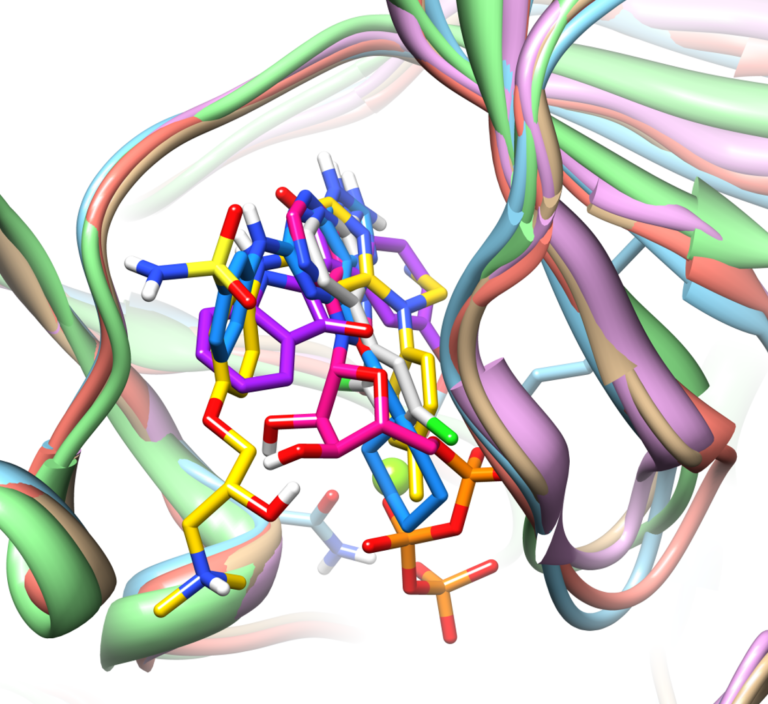
Knowledge-guided docking: accurate prospective prediction of bound configurations of novel ligands using Surflex™-Dock
We present an approach that uses structural information known prior to a particular cutoff-date to make predictions on ligands whose bounds structures were determined later. The knowledge-guided docking protocol was tested on a set of ten protein targets using a total of 949 ligands.
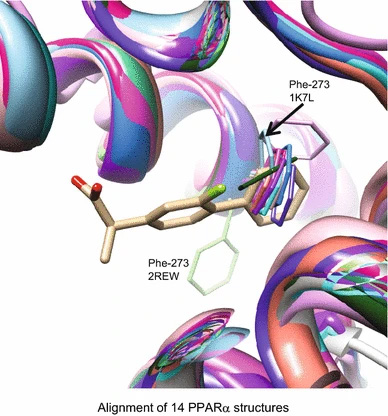
Chemical and protein structural basis for biological crosstalk between PPARα and COX enzymes
Here we present an analysis of novel drug/target predictions, focusing on those that were not obvious based on known pharmacological crosstalk.
Protein function annotation by local binding site surface similarity
This paper demonstrates how the Surflex-PSIM method can help investigate hypotheses around protein function in cases where function cannot be determined by sequence similarity.
