What’s the importance of cytochrome P450 metabolism?
Why focus on cytochrome P450 enzymes? CYPs are a ubiquitous superfamily of heme-containing monooxygenases responsible for approximately 70–80% of observed…

Why focus on cytochrome P450 enzymes? CYPs are a ubiquitous superfamily of heme-containing monooxygenases responsible for approximately 70–80% of observed…
Benefits of continuous integration to use up-to-date data in model building Cerella has the ability to work with and learn…

Buying software for your company can be a challenge. Every organisation does things differently, and there is often no handbook…

StarDrop — A Swiss Army knife for drug discovery It’s designed to fit right in with the other tools you…

At a very basic level, this means that StarDrop supports loading data from many different standard file formats (SDF, MOL2,…

Is AI-guided drug discovery faster and cheaper? The evidence for this is, by definition, anecdotal. No one runs the same…

What’s the purpose of a predictive model? What’s the value of predictive models for drug discovery? Most of the undergraduate…

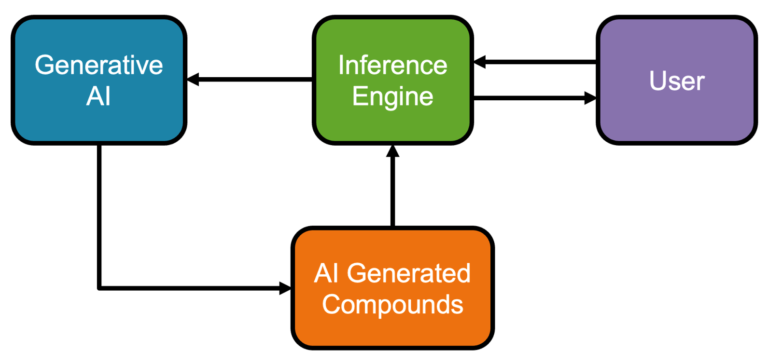
The role of generative chemistry in drug discovery A key difficulty in finding new drugs is the sheer size of…

Learn how to evaluate StarDrop with this step-by-step guide from Optibrium. Discover key features, trial objectives, and how to set up a successful evaluation of this powerful drug discovery platform

Discover which metabolite prediction software is best for your needs in this comprehensive guide from Optibrium. Compare top tools like Meteor Nexus, MetaSite, and StarDrop to make informed decisions for drug metabolism prediction

We’re often asked, “What’s the difference between QSAR and imputation models?”, so I’m going to explain how the methods differ, their advantages and disadvantages, and when each approach is applicable.

How number of users affect drug discovery software costs The number of people who need access to the platform is…

We recently published a case study with Amazon Web Services, detailing how we were able to scale our StarDrop platform…

Nearly all computational methods in the CADD field depend on parameters whose values are derived from various types of experimental…

If your current software has hidden costs, performance that can’t keep pace, poor support, or limited visualisation options, it might be time for a change. The good news is, switching to StarDrop is easier than you may think and this guide will walk you through every step.
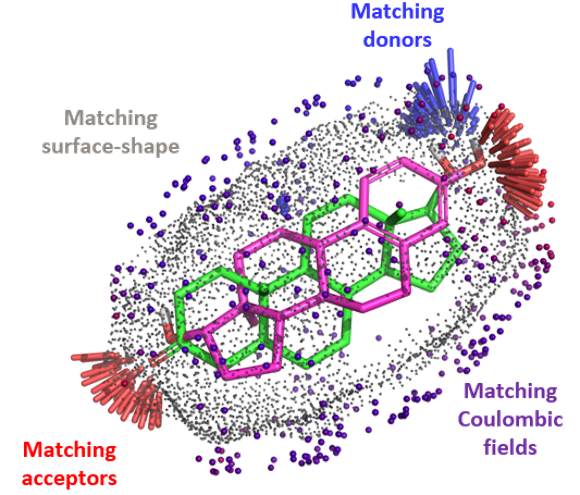
The QuanSA method To define a ‘pocket field’, an initial alignment of all training molecules is constructed and function parameters…
Pairing AI with human expertise We present a novel AI compound optimisation system, designed to include human oversight as a…