When using computational tools in drug discovery, what you can do depends on what you have available.
If you don’t have a target protein structure, there a range of ligand-based methods to use, such as similarity-based virtual screening and quantitative structure-activity relationships (QSAR), that leverage data from known active ligands.
However, advances in structural biology and protein structure prediction have made high-quality 3D structures more accessible. If you’re fortunate enough to have this for your project, you now have access to an additional set of powerful structure-based approaches.
But how do you make the most of this expanded toolkit?
In our recent article in Innovations in Pharmaceutical Technology, we explore the principles, techniques, and applications of structure-based and ligand-based methodologies, as well as how taking a combined approach can further enhance your drug discovery:
Better together: Why should you combine ligand-based and structure-based methods in your workflow?
Combining both approaches adds value as they capture different but complementary information:
Structure-based methods provide detailed information about specific protein-ligand interactions, including hydrogen bonds, hydrophobic contacts, and binding pocket geometry.
Ligand-based methods infer critical binding features from known active molecules, excel at pattern recognition and can generalise across chemically diverse compounds.
For instance, take ligand-based virtual screening and molecular docking. There are several ways that these can be combined to yield better results:
1. Sequential integration: Make the most of limited computational resources
One practical approach uses ligand-based methods as a quick filtering step before applying computationally intensive structure-based analysis. Large compound libraries are first screened using 2D/3D similarity against known actives or QSAR predictions. The most promising subset then undergoes molecular docking and binding affinity predictions.
This sequential approach improves efficiency, since more expensive and resource-intensive methods are used only on pre-filtered, high-potential compounds. The initial ligand-based screen can identify novel scaffolds early, offering chemically diverse starting points that docking can then analyse to optimise binding interactions. This approach works particularly well when time and resources are constrained, or when protein structural information emerges progressively.
Some pipelines run both methods simultaneously on the same compound library, generating independent rankings that can then be combined using consensus scoring.
Parallel scoring selects the top n% of compounds from both similarity rankings and docking scores. This produces a broader candidate set, increasing the chance of retrieving active compounds. Additionally, if docking scores are compromised by poor pose prediction or scoring function limitations, similarity-based methods may still recover actives based on known ligand features.
Hybrid scoring multiplies scores from each method to create a unified ranking. This favours compounds ranked highly by both approaches, prioritising specificity and increasing confidence in true positives.
Conclusion
In drug discovery, you need to make the most of what you have available.
Structure-based and ligand-based methods provide valuable, complimentary information on the same problem. By integrating them with consideration, you can reduce synthesis costs, improve hit rates, and increase your chances of identifying promising lead compounds.
Himani is a Principal Scientist at Optibrium, specialising in computational drug discovery. With a PhD in Bioinformatics and Computational Biology from the Indian Institute of Science, she has extensive experience in AI/ML techniques for modeling drug candidates, including small molecules and peptides. She is also involved in 3D ligand-based and structure-based drug design.
Before joining Optibrium, Himani completed a postdoctoral fellowship at the MRC Laboratory of Molecular Biology, focusing on cell reprogramming and transcriptomics.
We present a hybrid structure-guided strategy that combines molecular similarity, docking, and multiple-instance learning such that information from protein structures can be used to inform models of structure–activity relationships.
Systematic optimisation of large macrocyclic peptide ligands is a serious challenge. Here, we describe an approach for lead optimisation using the PD-1/PD-L1 system as a retrospective example of moving from initial lead compound to clinical candidate.
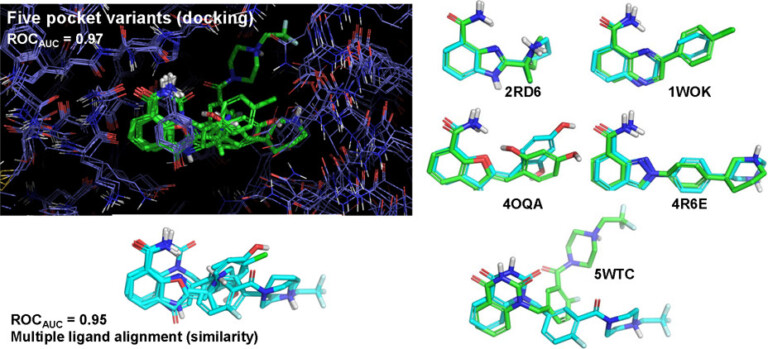
Using the DUD-E+ benchmark, we explore the impact of using a single protein pocket or ligand for virtual screening compared with using ensembles of alternative pockets, ligands, and sets thereof.