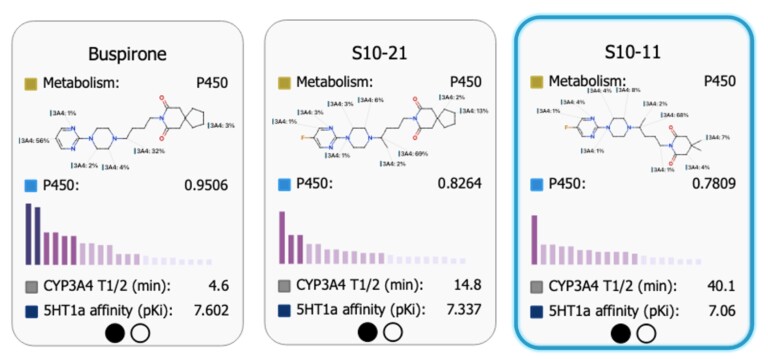
Shorter development cycle, bigger impact: What’s new in StarDrop™ 8.1
It feels like only yesterday that we announced the arrival of StarDrop 8. And here we are again. Though this time, that’s rather the point. Discovery teams need to iterate quickly, moving from hypothesis…