We’re often asked, “What’s the difference between QSAR and imputation models?”, so I’m going to explain how the methods differ, their advantages and disadvantages, and when each approach is applicable.
What are QSAR models?
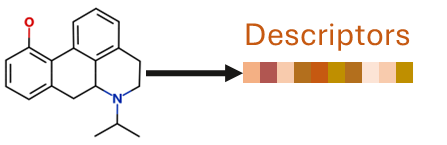
QSAR stands for Quantitative, Structure-Activity Relationship; in other words, QSAR models capture how differences between chemical structures relate to differences in their activities or other properties. They work by calculating features of a compound’s structure, or ‘descriptors’.
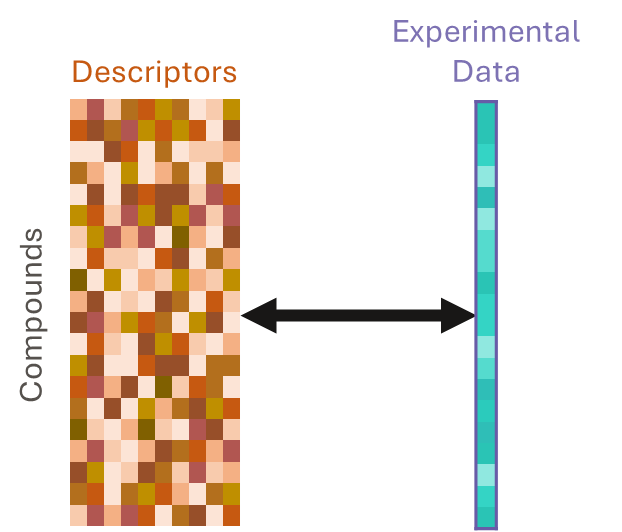
QSAR models are built using statistical or machine learning methods to find correlations between the descriptors of many compounds and their experimentally measured values of a property.
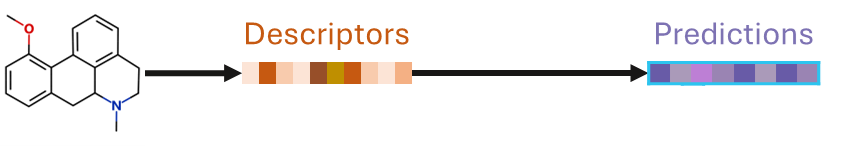
Once built, a QSAR model only needs the chemical structure of a new compound to predict its properties.
This is ideal for assessing many new compound designs to choose the best compounds to synthesise and test experimentally, for example, for virtual screening or in combination with generative chemistry methods.
Imputation models are quite different from QSAR
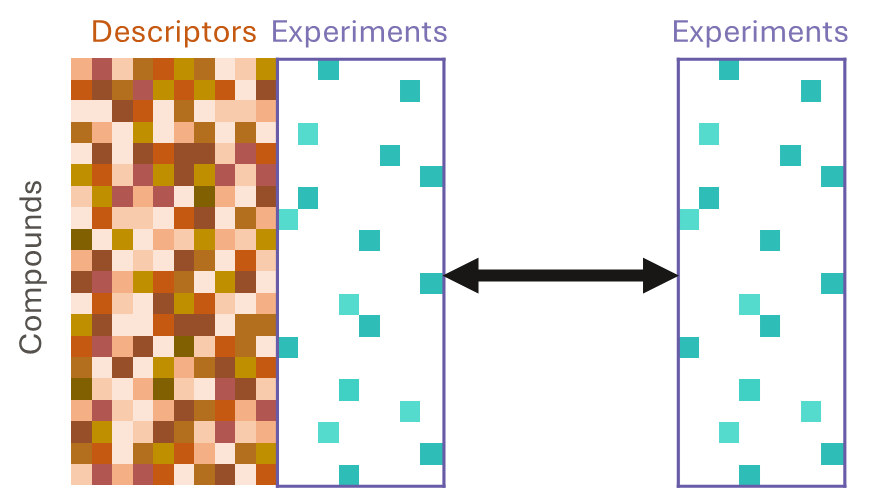
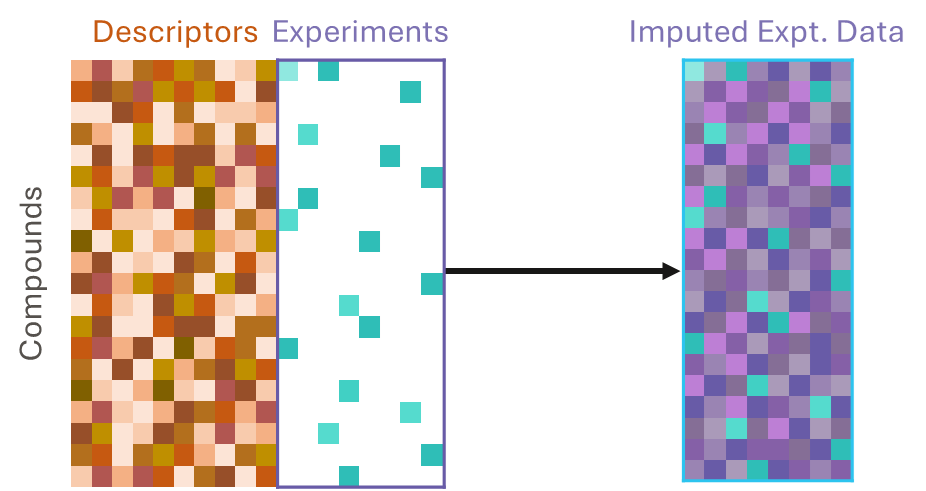
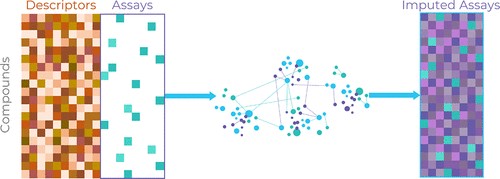
They use both a chemical structures and experimental data as inputs. They learn from correlations between compound descriptors and measured data and between measurements of different experimental endpoints. This approach can gain much more information than QSAR, even when data for many compounds and endpoints are missing; in other words, it can use sparse data.
To make predictions using an imputation model, we can input a compound’s structure and any data we have already measured to predict the results of experiments we have not yet run on that compound, thereby imputing, or filling in, the missing data.
And imputation can be applied to an entire matrix of compounds and data.
So, why is this relevant to drug discovery?
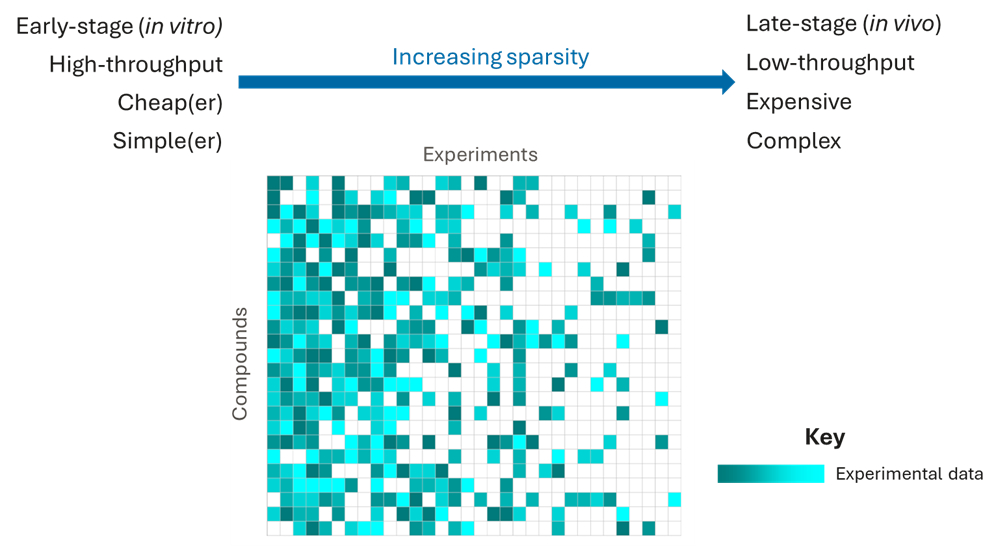
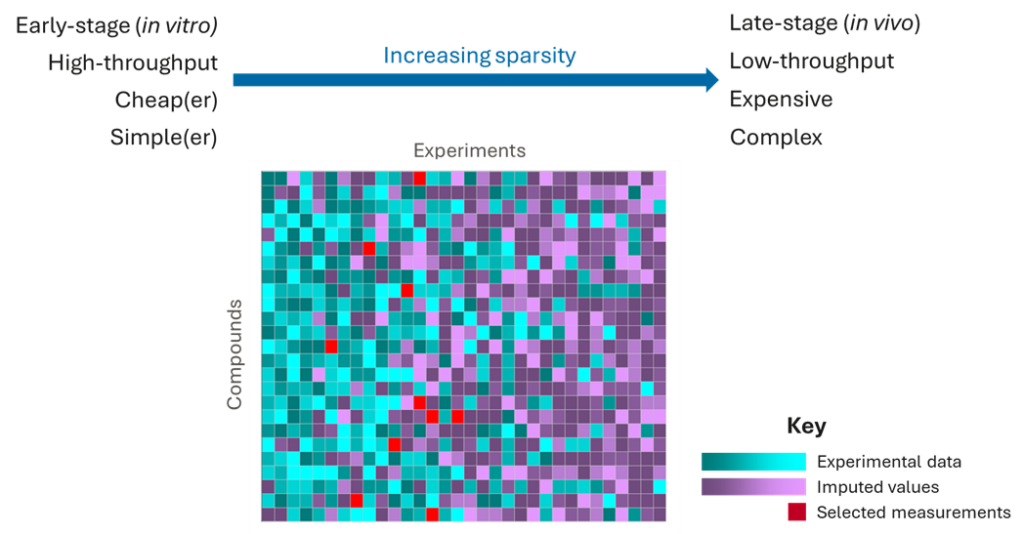
The data we have in drug discovery is very sparse. We typically have plenty of data for early, high-throughput, low-cost experiments, such as in vitro biochemical activities or simple physicochemical properties. But, as we progress downstream to more expensive, lower throughput experiments, such as phenotypic screens or in vivo studies, we have less and less data.
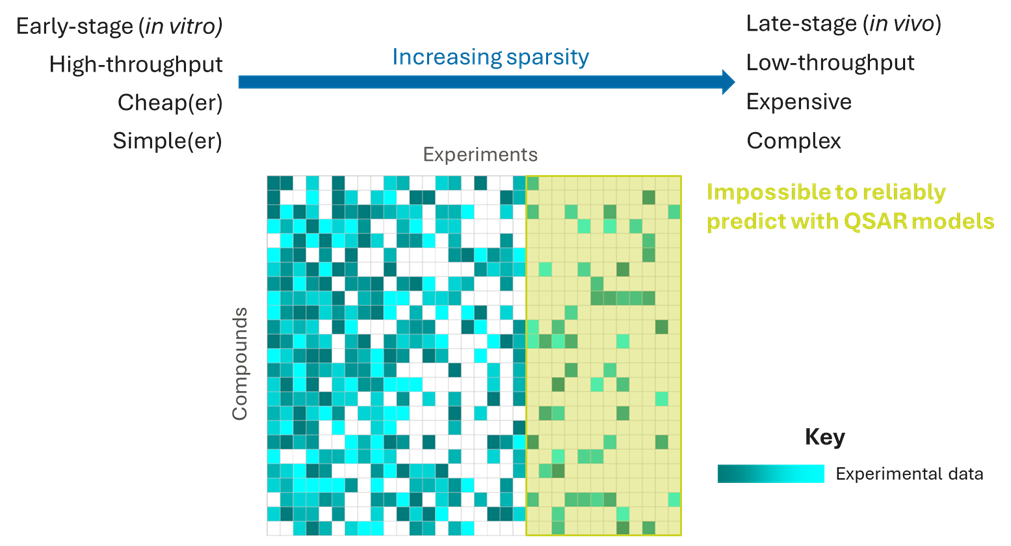
Of course, these later-stage experiments are the most valuable for selecting the best candidate drugs. But, these outcomes are impossible to predict using QSAR models because they are very complex, often being determined by multiple factors, including target activities, ADME and physicochemical properties, meaning that an enormous amount of data would be required to dissect these complex relationships with chemical structures. And, of course, these are the experiments for which we have the least data!
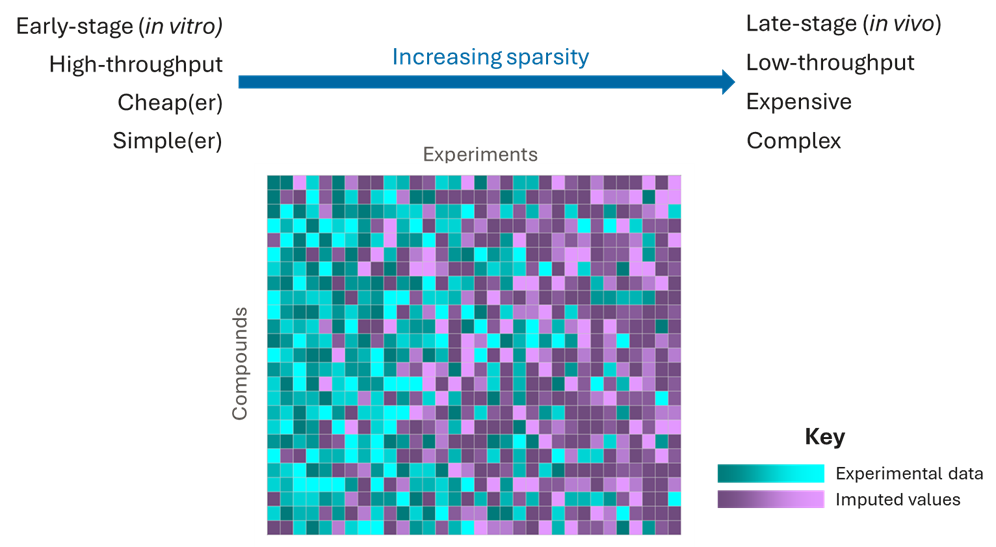
However, the data from the earlier experiments contain very valuable information with which to predict downstream experimental outcomes, and imputation methods can leverage these data to make much more accurate predictions than QSAR models by filling in the missing values.
That, in turn, enables us to make much more confident predictions of the best compounds to progress, saving time and money while improving the chances of delivering a high-quality candidate.
And it’s not only about making more accurate predictions.
Building an imputation model captures information about the relationships between different experimental endpoints, giving us additional valuable insights.
The relationships between experimental endpoints, revealed from the complex, sparse data by an imputation model, can help to identify mechanisms of action and adverse outcome pathways.
The model can also use this information to suggest the next experiments to perform that will generate the most valuable data to make even more accurate predictions. This ‘active learning’ approach maximises the return on investment for your experimental resources.
And, the model can identify measured values that are highly unlikely given all of the other data in your data set. These outliers reveal valuable new opportunities that you may have missed due to missing, uncertain or incorrect data, such as false negatives.
So, imputation models are most applicable when you have a small amount of experimental data for your compounds and want to use these to make the best decisions to progress from concept to candidate faster and cheaper.
Interested in building your own imputation and QSAR models?
Cerella automatically builds and updates both imputation and QSAR models, so you can benefit from both methods and ensure that you make decisions based on the latest information.
Matt holds a Master’s in Computation from the University of Oxford and a PhD in Theoretical Physics from the University of Cambridge. He led teams developing predictive ADME models and advanced decision-support tools for drug discovery at Camitro (UK), ArQule Inc., and Inpharmatica. In 2006, he took charge of Inpharmatica’s ADME business, overseeing experimental services and the StarDrop software platform. After Inpharmatica’s acquisition, he became Senior Director of BioFocus DPI’s ADMET division and, in 2009, led a management buyout to establish Optibrium.
In this ebook we demonstrate our deployable AI discovery platform, Cerella™. Browse real-world stories of success from our collaborations with AstraZeneca, Genetech, Takeda Pharmaceuticals, Constellation Pharmaceuticals and many more.