Avoid the hype – how to successfully implement AI in drug discovery
AI has the potential to transform discovery. However, to ensure real impact, there are several practicalities that organisations must consider…

AI has the potential to transform discovery. However, to ensure real impact, there are several practicalities that organisations must consider…
Ann and Ajay discuss the science behind and applications of the eSim molecular similarity method, a ligand-based drug design approach which considers surface-shape, electrostatics, and directionally sensitive hydrogen-bonding when comparing two molecules.
This peer-reviewed paper in Xenobiotica describes a new method to determine the most likely experimentally-observed routes of metabolism and metabolites based on our WhichP450™, regioselectivity and new WhichEnzyme™ model.
We explore the exciting new features in the latest release of StarDrop, built to elevate your drug discovery projects. These include the all-new Metabolism module; high performance virtual screening; additional workflow improvements

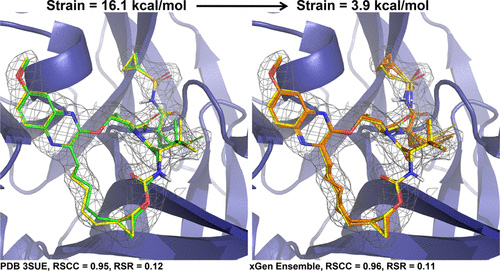
We show that the distribution of expected global strain energy values is dependent on molecular size in a superlinear manner. The distribution of strain energy follows a rectified normal distribution whose mean and variance are related to conformational complexity.
Now, watch Matt Segall, PhD, CEO at Optibrium, as he introduces a real world case study where we applied deep learning to guide a project, in which potential compounds were displaying good activity profiles but the team wanted to improve their PK profile to achieve better efficacy.
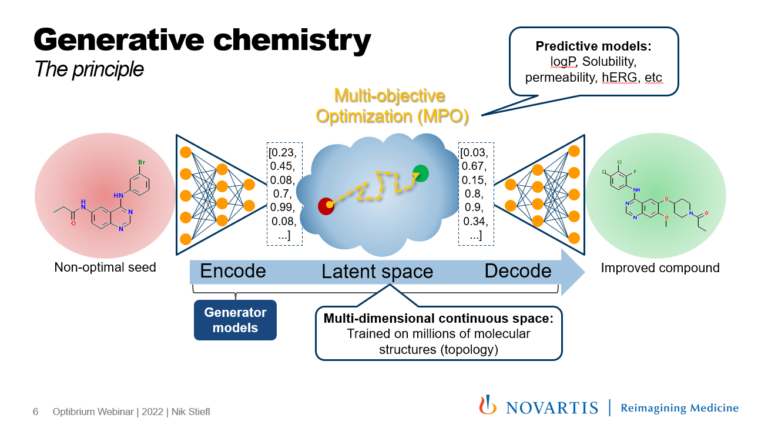
Have advances in AI and deep learning reached a threshold whereby generative chemistry methods are redefining drug design? This webinar…
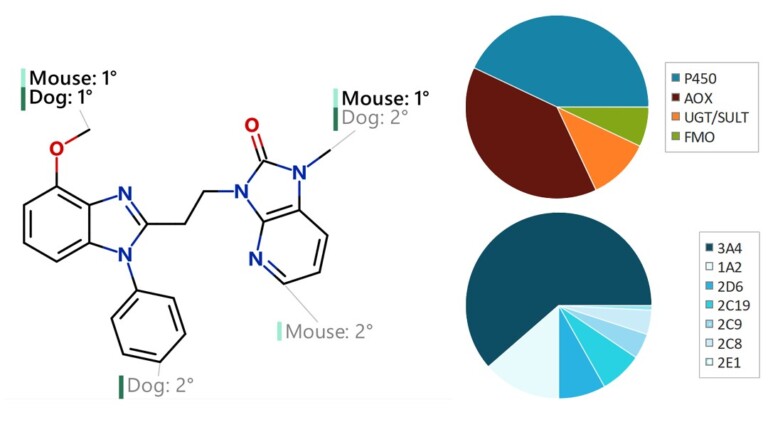
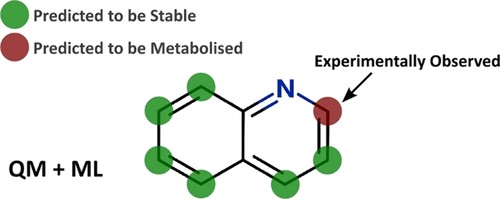
This paper describes the prediction of the regioselectivity of metabolism by AOs, FMOs and UGTs for humans and CYPs for three preclinical species.
In this webinar, learn about Cerella’s unique AI methods, see examples of its successful application throughout the drug discovery process and watch a demonstration of how CDD Vault and Cerella connect to seamlessly integrate with your workflows.
In this webinar, we examine the effective use of QSAR modelling in drug discovery and discuss a variety of pain points for medicinal chemists in knowing when a model can be trusted and how to avoid common pitfalls.
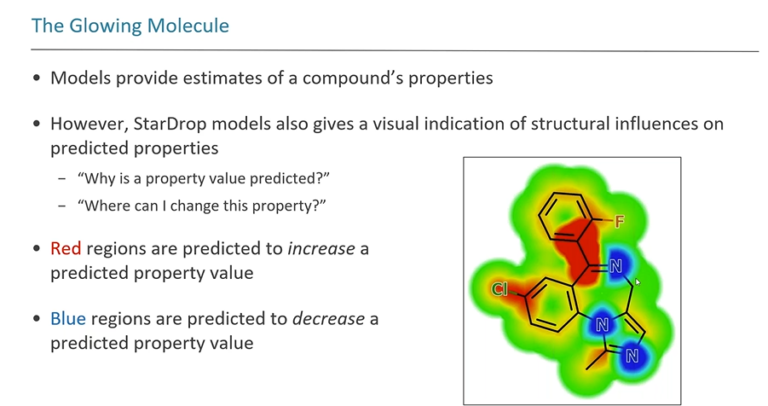
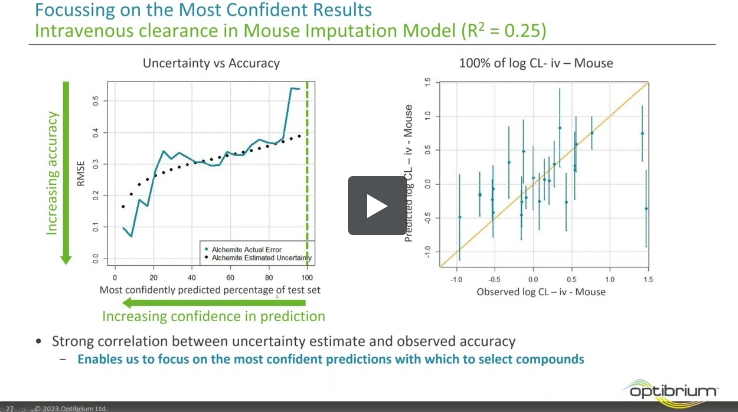
In this webinar, we explore the highlights of collaborative project results that demonstrate how every phase of the drug discovery process can be radically improved by applying proven AI technology. Providing scientists with insights on which to base decisions can identify valuable new opportunities and reduce the time and cost of AI drug discovery cycles.
We review case studies from collaborations with Constellation Pharmaceuticals, AstraZeneca, Genentech, the University of Dundee and Takeda Pharmaceuticals to validate the impact of applying AI to experimental data and illustrate dramatic improvements to their project outcomes.
Join Samar Mahmoud and Matt Segall for this fascinating deep dive into the revolution that AI is bringing to the challenges of sparse and noisy drug discovery data.
In this webinar, we demonstrate how Inspyra™ creates a seamless blend of your expertise and unique AI that fits naturally within your workflow. It helps you to rigorously explore many optimisation strategies and quickly identify high-quality compounds for your projects.

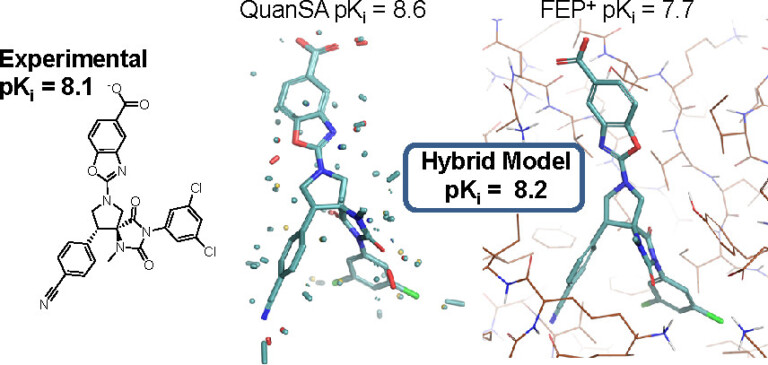
We present results on the extent to which physics-based simulation (exemplified by FEP+) and focused machine learning (exemplified by QuanSA) are complementary for ligand affinity prediction.
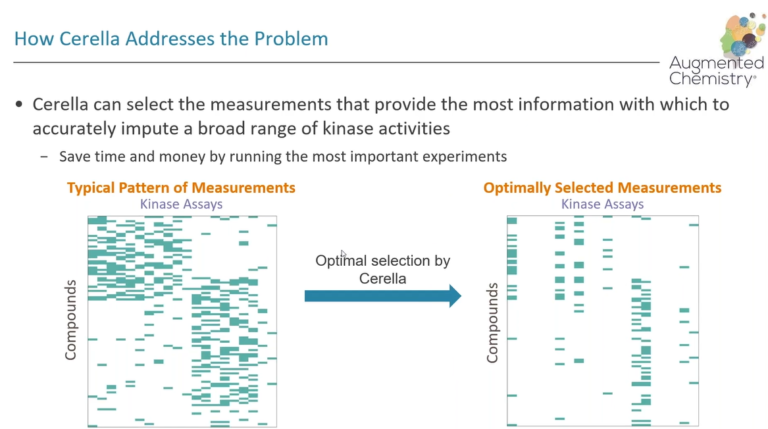
In this webinar, we demonstrate how Augmented Chemistry®, a unique deep learning method, can learn from higher throughput data together with limited panel data to provide high-quality imputations for sensory properties.
Why have generative chemistry methods been unable to redefine modern drug discovery and compound idea generation?’ In this session we shed light on a typical shortcoming of generative methods related to prioritising promising over unsuitable directions for exploration.
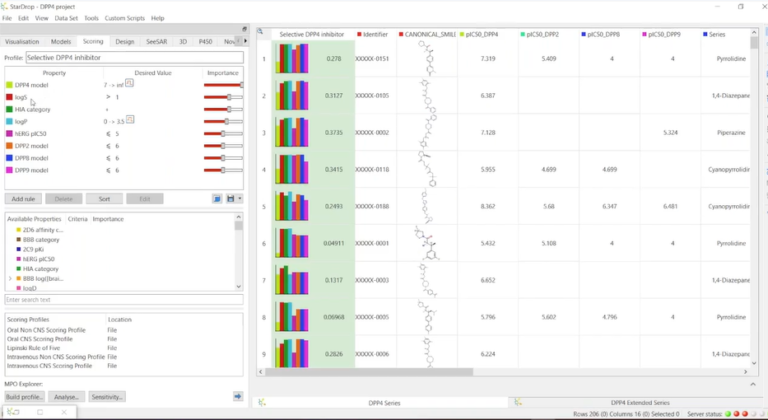
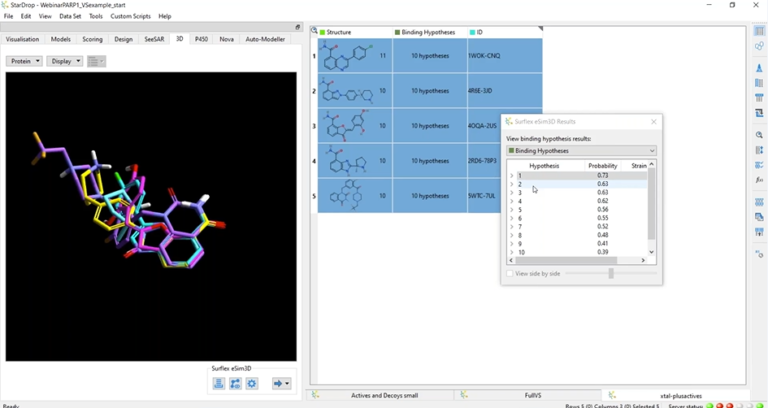
In this webinar, we demonstrate intuitive workflows for 3D ligand-based drug design
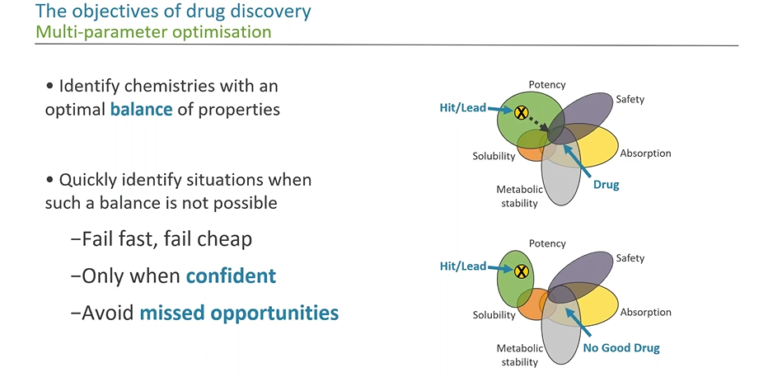
In this webinar, we look at how we can use data visualisation in an impactful and effective way to communicate many dimensions of information. We illustrate some of the ways that we can achieve this and discuss visual methods to guide our decisions in drug discovery.
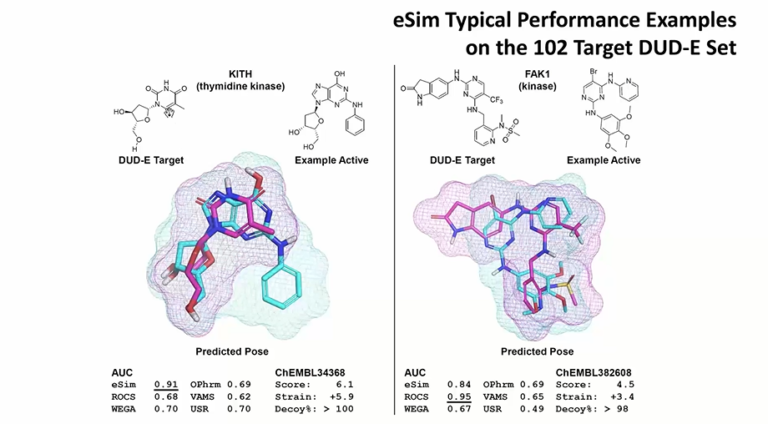
In this webinar, we present eSim3D, a novel ligand-based drug design approach based on electrostatic-field and surface-shape similarity coupled with unique conformational search capabilities, offering unprecedented accuracy and performance.
In this webinar, we described the generation and validation of a ‘global’ model using deep learning imputation on a data set of 300,000 compounds and 500 experimental endpoints, targeting global health indications.
We demonstrated how this global model can be applied to individual optimisation projects, offering improved compounds design performance over ‘local’ project-specific models by learning across a broad chemical diversity.