In drug discovery, we tend to operate with a familiar simplification that molecules, both ligands and targets are static. A ligand fits into the binding pocket like a key into a lock, and our computational methods treat them accordingly. While this assumption may be sufficient for early-stage high-throughput experiments, it misrepresents how molecules behave. In reality, both ligands and proteins are dynamic and flexible. They twist, bend, flip, and sample a range of 3D conformations in solution. We can’t ignore this as the conformation of a molecule can impact its on- and off-target potency, ADMET properties, and other physicochemical characteristics.
Conformational ensembles enable us to sample the full distribution of geometries, providing a more accurate representation of how ligands exist in solution. This in turn improves the accuracy of our computational predictions of binding modes and affinities.
In this post, we will explore why conformational ensembles are not just useful, but often essential, particularly as the size and complexity of a molecule increases, and we move away from traditional small molecules, into more flexible, structurally complex chemical space, which is mostly the focus of this post.
What are conformational ensembles?
A conformational ensemble is a collection of the different 3D shapes a molecule can adopt in solution, weighted by their relative population. Rather than representing a molecule as a single, static structure, an ensemble captures its full flexibility. Each conformer exists at a specific energy level determined by molecular strain, steric interactions, and stabilising interactions such as hydrogen bonds and disulphide bonds (for bigger molecules and proteins).
High quality conformational sampling identifies not only the global minimum energy conformation but also a biologically active conformation, a conformation in which molecules bind to their targets. In high-quality conformational sampling, the goal is not just to obtain a global minimum, but to also map the entire low energy space. By modelling this ensemble rather than a single shape, we move closer to how molecules actually behave. This has profound implications for both structure-based and ligand-based methods to accurately predict binding modes and affinities or activities.
How do conformational ensembles improve predictive modelling?
Higher accuracy in molecular docking
Molecular docking aims to predict how ligands will fit into the binding site of the target protein and rank their binding potential. It relies on an accurate representation of ligand flexibility to predict binding poses. By starting with a diverse low-energy ensemble of ligand conformations, we increase the chances of finding a geometry that is complementary to the protein pocket. This improves the robustness of pose prediction and can reveal binding modes that would be missed when using only a single, low-energy conformation. This is especially important for flexible ligands like macrocycles or peptides. Proteins are often considered rigid for simplification by various docking tools; however, some tools go a step further by incorporating protein ensembles to capture some aspect of pocket flexibility.
An example of conformational sampling for a macrocyclic peptide – Aureobasidin A. As the size and flexibility of a macrocycle increases, the number of accessible conformers grows exponentially due to the increased degrees of freedom. This makes exhaustive conformational sampling not only challenging, but also critical for accurate docking.
Better performance in ligand-based virtual screening
n 3D ligand-based approaches, molecules are compared by shape, electrostatics, or pharmacophoric features. Incorporating conformational ensembles in similarity searches accounts for ligand flexibility and allows us to perform similarity calculations more accurately by acknowledging that a molecule’s bioactive conformation may be one possibility among many. If you only rely on a single conformation, you are effectively taking a static snapshot of something that is inherently dynamic. This will become limiting in screening chemically diverse molecules that can adopt structurally equivalent pharmacophoric arrangements, but in an alternative conformation. These are the conformations that actually enable key interactions i.e., H-bonding, electrostatic, or hydrophobic contacts with the target protein.
This is particularly valuable when searching for bioisosteres or performing scaffold hops, where the goal is to discover new chemical cores that can reproduce the key interactions of known actives in 3D. If such similarity only emerges in a relatively higher energy conformer, which is still biologically accessible, a single structure comparison will miss it entirely. Hence, incorporating conformational ensembles for ligand-based screening results in a much richer and more diverse set of potential hits.
Integration of biophysical constraints
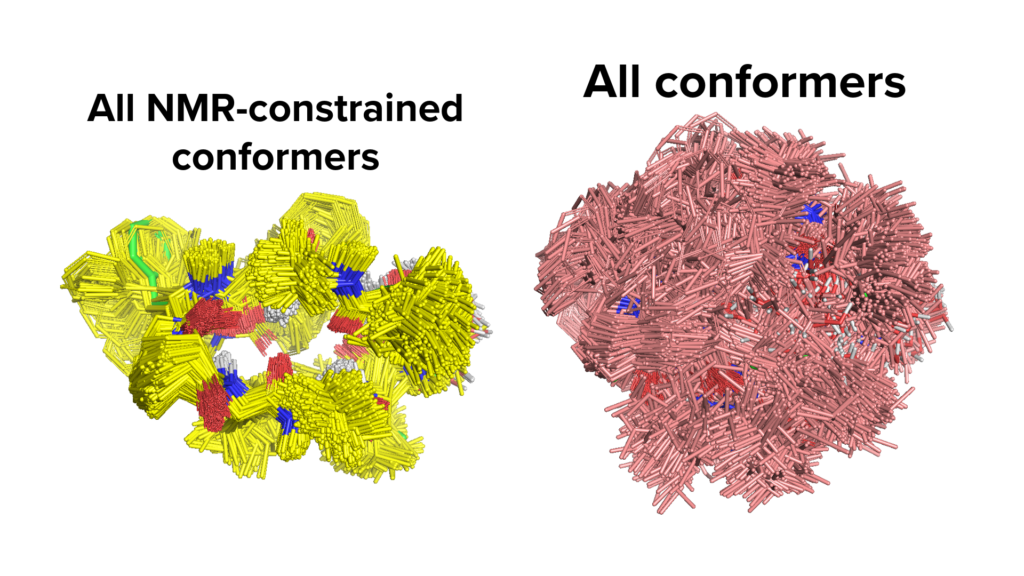
As the size of the molecules increases, the number of accessible conformers grows exponentially due to the increased degrees of freedom. For such molecules, obtaining a meaningful conformational ensemble is often not trivial and this is particularly challenging for bigger macrocycles, like peptidic ones. However, the search space for such molecules can be narrowed down if one has got some biophysical data e.g., restraints from NMR, X-ray or CryoEM. Biophysical constraints can help bridge the gap between purely theoretical sampling and conformations molecules adapt in physiological environment and increases the likelihood of finding the bioactive conformation within the pool. We can refine the ensemble by incorporating such biophysical constraints into our confirmational sampling.
An example of conformational sampling for Aureobasidin A with (left) and without (right) NMR constraints. Such biophysical restraints can help restrict the search space and increase the likelihood of obtaining a bioactive conformation within the pool.
Improved property prediction for complex molecules
3D structure of molecules can have significant impact on their properties and stability of the bioactive conformation influences their potency, solubility, permeability etc, making property prediction highly dependent on conformation. These properties are influenced not just by a molecule’s lowest energy but by the range of conformers it can adopt. Therefore, it becomes critical to reliably generate relevant conformers. As our chemical matter moves beyond traditional Ro5 small molecules into larger more flexible molecules like macrocycles, peptides, and other bRo5 scaffolds, the limit of single conformer modelling becomes increasingly apparent. Conformational ensembles offer a more realistic and predictive foundation for modelling properties that are highly conformation-dependant.
Improved x-ray density structure fitting
Anyone who has worked with structures deposited in PDB would not be surprised by the fact that ligand coordinates are often poorly fitted into the X-ray density. This issue is particularly pronounced for macrocycles (both peptidic and non-peptidic). Yet these deposited conformations are frequently used as gold-standard for downstream analyses, from docking benchmarks to strain energy calculations, which can lead to misleading conclusions. For example, ligand strain energy, defined as the difference in the energy between the bound and unbound conformation is often overestimated when calculated directly from PDB-deposited ones.
Using conformational ensembles to fit into X-ray electron density, we can avoid such pitfalls. Rather than forcing a single conformer to explain all the density, such ensembles can provide much better fits that respect the experimental data. This is particularly valuable for complex molecules like macrocycles where this approach leads to more accurate geometry, better interpretation of ligand interactions, and reliable strain energy estimates.
ForceGen: Fast and accurate conformer sampling from small molecules to macrocycles
ForceGen is your 3D chemical toolkit. Its template-free method systematically explores accessible conformational space through 3D physical manipulations, to generate ensembles of diverse and low-energy conformers. It can evaluate complex macrocycles in minutes, with the option of applying NMR constraints to increase search efficiency.
Starting from just a 2D SDF or SMILES, it can generate a pool of up to 1000 conformers that can integrate with the full BioPharmics 3D modelling suite:
Surflex™-Dock for fully automated molecular docking
eSim for high-performance 3D virtual screening
QuanSA for ligand-based binding affinity prediction.
xGen: Refining conformational ensembles with x-ray electron density maps
xGen is our novel method for real-space refinement and de novo fitting of ligand ensembles into X-ray density maps. It produces chemically sensible conformers with low strain energy, and is applicable to small molecules and complex macrocycles.
XGen has been proven to yields superior fit to X-ray density than standard fitting approaches, and is accessible to non-crystallographers and as part of crystallographic workflows.
About the author
Himani Tandon, PhD
Himani is a Principal Scientist at Optibrium, specialising in computational drug discovery. With a PhD in Bioinformatics and Computational Biology from the Indian Institute of Science, she has extensive experience in AI/ML techniques for modeling drug candidates, including small molecules and peptides. She is also involved in 3D ligand-based and structure-based drug design.
Before joining Optibrium, Himani completed a postdoctoral fellowship at the MRC Laboratory of Molecular Biology, focusing on cell reprogramming and transcriptomics.
We present an approach that uses structural information known prior to a particular cutoff-date to make predictions on ligands whose bounds structures were determined later. The knowledge-guided docking protocol was tested on a set of ten protein targets using a total of 949 ligands.