Optibrium Widens Access to Industry-Leading Docking Method
New PyMOL interface extends access to Optibrium’s structure-based design method Surflex™-Dock CAMBRIDGE, UK, 04 February 2025 – Optibrium, a leading…
You might be sceptical about ligand-based QSAR approaches; many researchers are. We discussed why there has been a lack of confidence in traditional QSAR methods to predict binding affinity in our recent Bio-IT World article.
One factor is that these statistical models typically use inputs that bear little or no connection to the underlying physics of binding. They also often struggle to predict activities for structurally novel molecules that differ from the training set, which is precisely what we want to do in drug discovery.
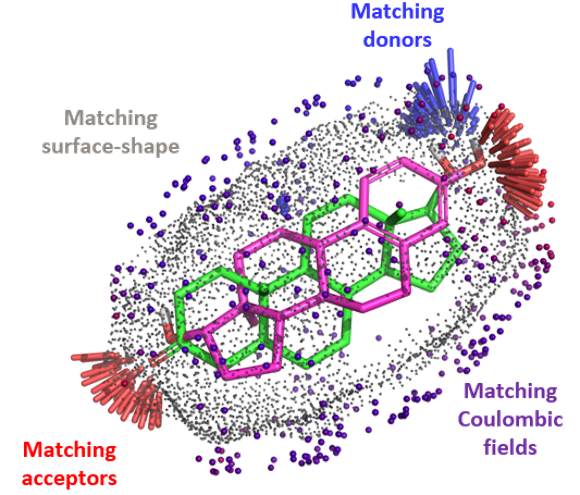
QuanSA (Quantitative Surface Field Analysis) was developed specifically to overcome these limitations. Rather than treating affinity prediction as a purely statistical problem, QuanSA builds virtual binding pockets directly from your ligand data. They represent the physical reality of protein-ligand interactions without requiring the actual protein structure.
What makes this possible is multiple-instance machine learning. QuanSA automatically aligns your ligands in 3D space, with the model evolving as it learns from the ligand poses. Unlike traditional QSAR, where alignment is fixed at the start, QuanSA dynamically identifies and refines optimal poses as its parameters develop, learning both structure and physical interactions simultaneously.
This enables QuanSA to account for what actually drives binding:
By modelling these physical interactions rather than relying on statistical correlations alone, QuanSA can generalise to structurally diverse molecules, including scaffold hops, which is exactly what you need when exploring new chemical space.
QuanSA delivers comparable accuracy to FEP whilst being roughly 1,000 times less computationally expensive. Once you’ve built a model (which takes hours, not weeks), scoring new compounds happens in seconds. The model has already learned the essential physics of binding from your training data, so evaluating how well a new molecule fits into the learned pocket field, accounting for shape, hydrogen bonding, electrostatics, and strain, is computationally straightforward.
In addition to being able to make accurate binding affinity predictions without waiting for a target structure, this enables you to:
In collaboration with Bristol Myers Squibb, we found improved affinity prediction with QuanSA in LFA-1 inhibitor lead optimisation.
This project focused on identifying orally available small molecules targeting the LFA-1/ICAM-1 interaction, which modulates immune responses. Structure-activity data from these compounds were split into chronological training and test datasets for QuanSA and FEP+ affinity predictions.
Each individual method showed similar levels of high accuracy in predicting pKi. Furthermore, a hybrid model that averaged predictions from both approaches performed even better. QuanSA and FEP+ make uncorrelated errors, so this work shows how you can take advantage of both when structures are available
The QuanSA plugin for PyMOL makes these predictions more widely accessible. The plugin’s clear visualisations identify the key interactions that drive molecular affinity, providing essential insights to design even more potent compounds.
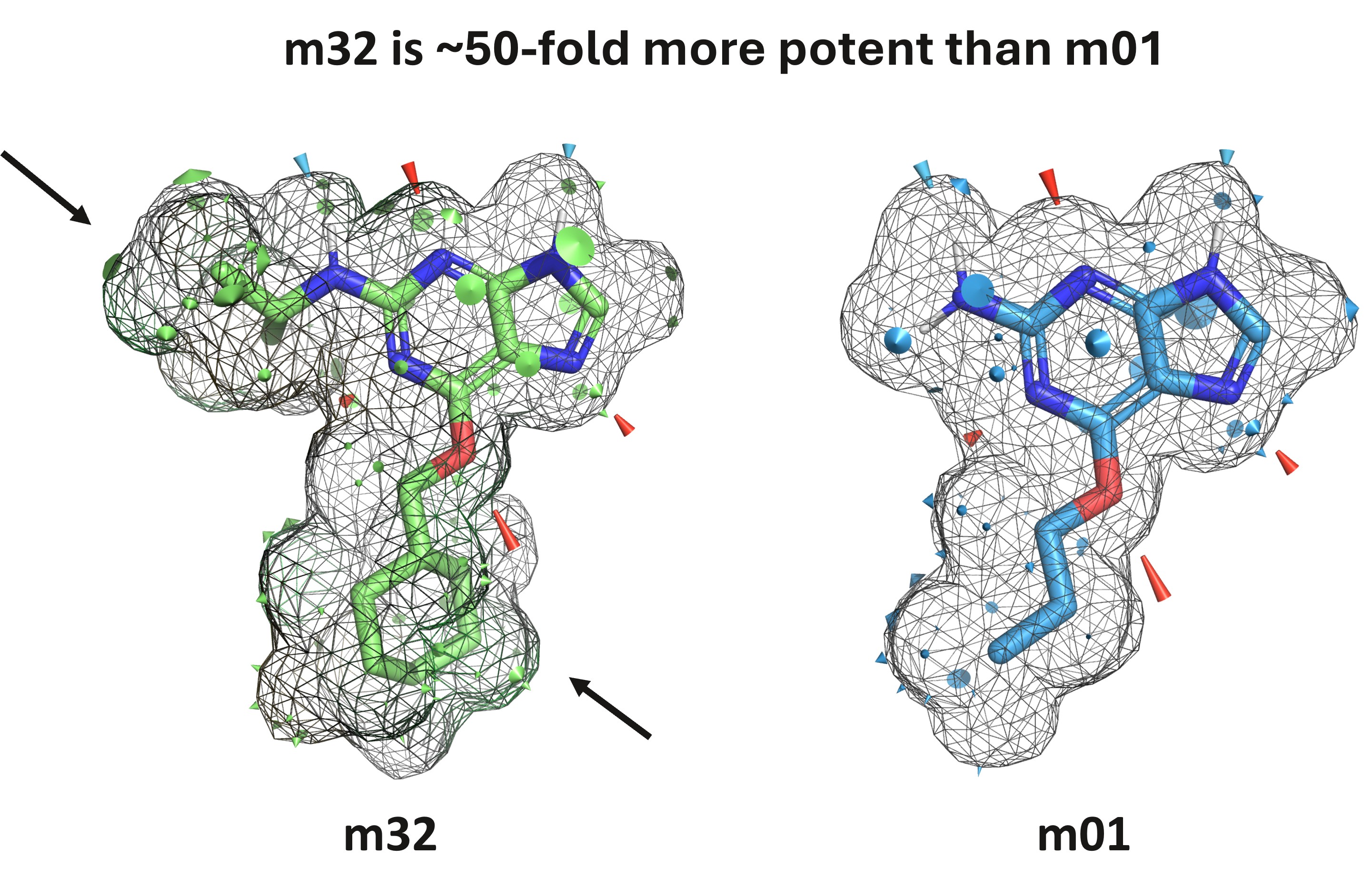
The absence of a protein structure doesn’t have to stop your affinity prediction work. Whilst structure-based methods like FEP are powerful when you have high-quality structures, QuanSA provides a reliable alternative that’s fast enough for high-throughput screening, accurate enough to guide design decisions, and transparent enough to build confidence in predictions.
Want to try it out for yourself? QuanSA is part of our BioPharmics platform, which provides industry-leading 3D methods for structure and ligand-based design. Fill the form below to learn more.
Simply complete the form now for a free demo and see how BioPharmics fits your unique research needs.
Himani is a Principal Scientist at Optibrium, specialising in computational drug discovery. With a PhD in Bioinformatics and Computational Biology from the Indian Institute of Science, she has extensive experience in AI/ML techniques for modeling drug candidates, including small molecules and peptides. She is also involved in 3D ligand-based and structure-based drug design.
Before joining Optibrium, Himani completed a postdoctoral fellowship at the MRC Laboratory of Molecular Biology, focusing on cell reprogramming and transcriptomics.

New PyMOL interface extends access to Optibrium’s structure-based design method Surflex™-Dock CAMBRIDGE, UK, 04 February 2025 – Optibrium, a leading…
The QuanSA method To define a ‘pocket field’, an initial alignment of all training molecules is constructed and function parameters…
Binding affinity prediction continues to be a challenge in computer-aided drug design, especially in the absence of a high-quality target…
