Introduction
Using the integrated set of computational methods within the BioPharmics™ Platform, macrocycles can be effectively modelled for lead optimisation. Here, we present a retrospective case study involving optimisation of a macrocyclic inhibitor of the PD-1/PD-L1 interface that was discovered through mRNA library screening. We demonstrate acceleration of lead optimisation based on NMR data on the lead compound and a single crystal structure of the target protein using a combination of deep conformational search, molecular docking, and careful estimation of bound ligand strain. Additionally, a ligand-based approach for predicting bound poses was also effective in prioritising lead-compound analogues.
Thousands of analogues were synthesised and tested to develop the clinical candidate, BMT-174900, a macrocyclic peptide inhibitor of PD-L1/PD-1 interaction, starting from the lead compound Pep-01.
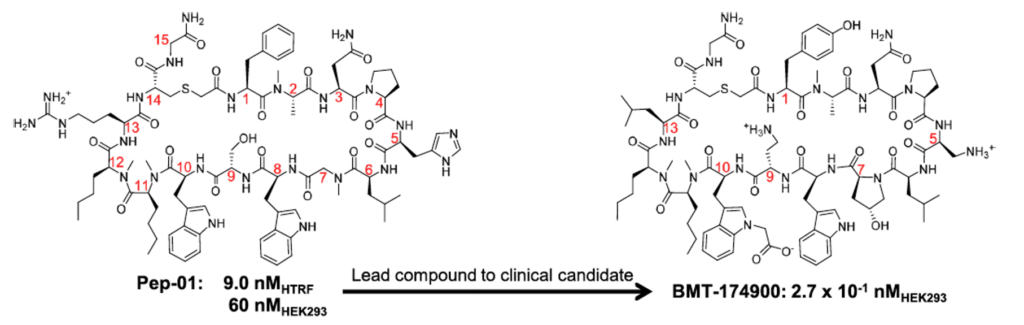
Objective
Develop an efficient approach to advance a lead macrocyclic peptide to a clinical candidate – faster, cheaper, and with accuracy.
This was achieved by integrating biophysical data from the lead peptide with practical, efficient computational methods within the BioPharmics suite, in collaboration with BMS, to prioritise compounds for synthesis.
Dataset and methods
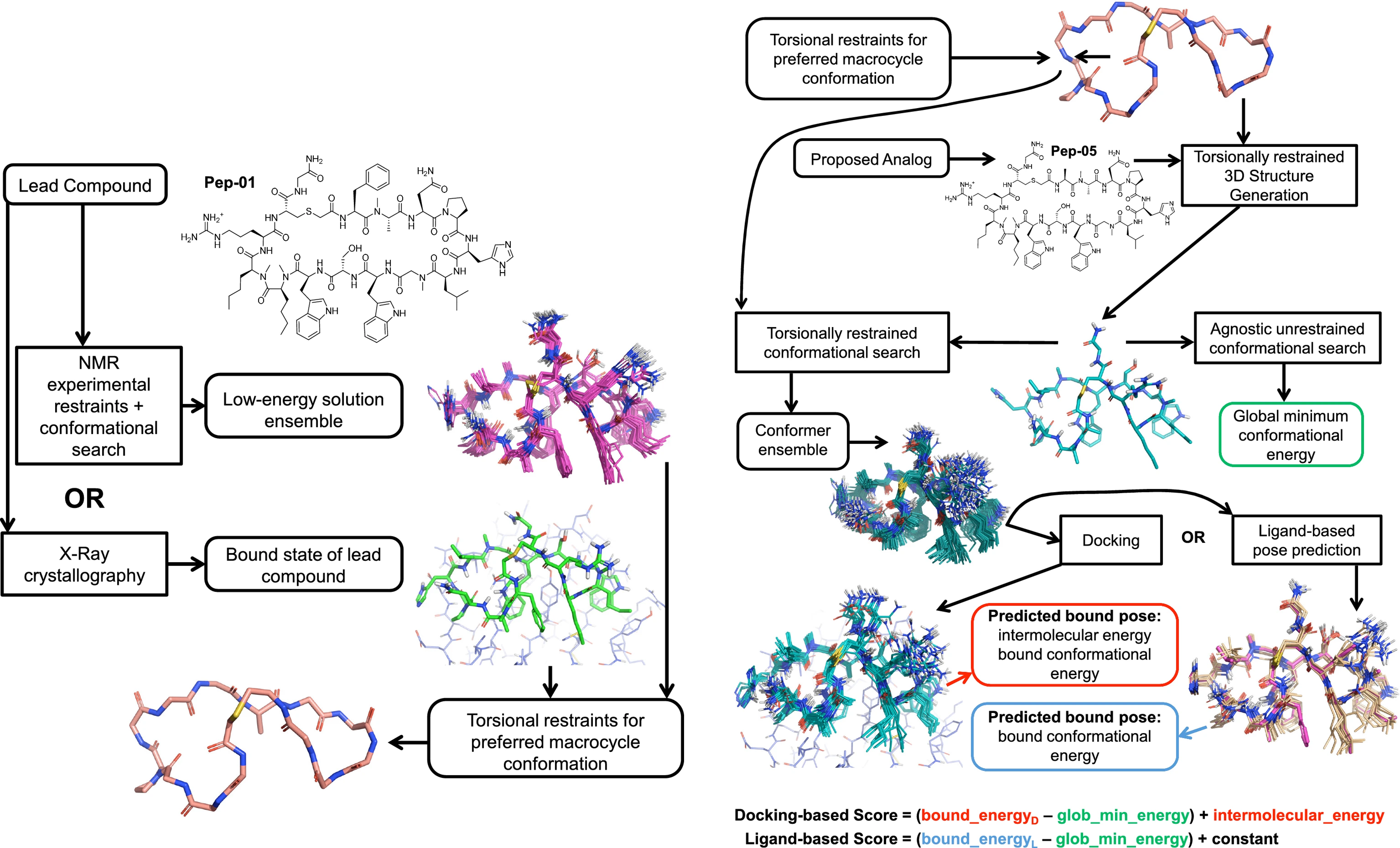
72 macrocyclic peptides, including the lead and optimised peptides, from the BMS patent disclosure1, each with associated IC50 values.
- NMR restraints: 50 proton-proton distance restraints, 115 involving chemically equivalent protons, and 6 torsional restraints (1 omega, 5 psi).
- ForceGen: Template-free, force-field-based conformational search with NMR-restraints2 using the fgen_deep approach for enhanced sampling
- Surflex™-Dock: Flexible-ligand, ensemble docking leveraging prior bound ligand knowledge for accurate pose prediction3.
- eSim: Electrostatic-field and surface-shape similarity-based poses4.
Results
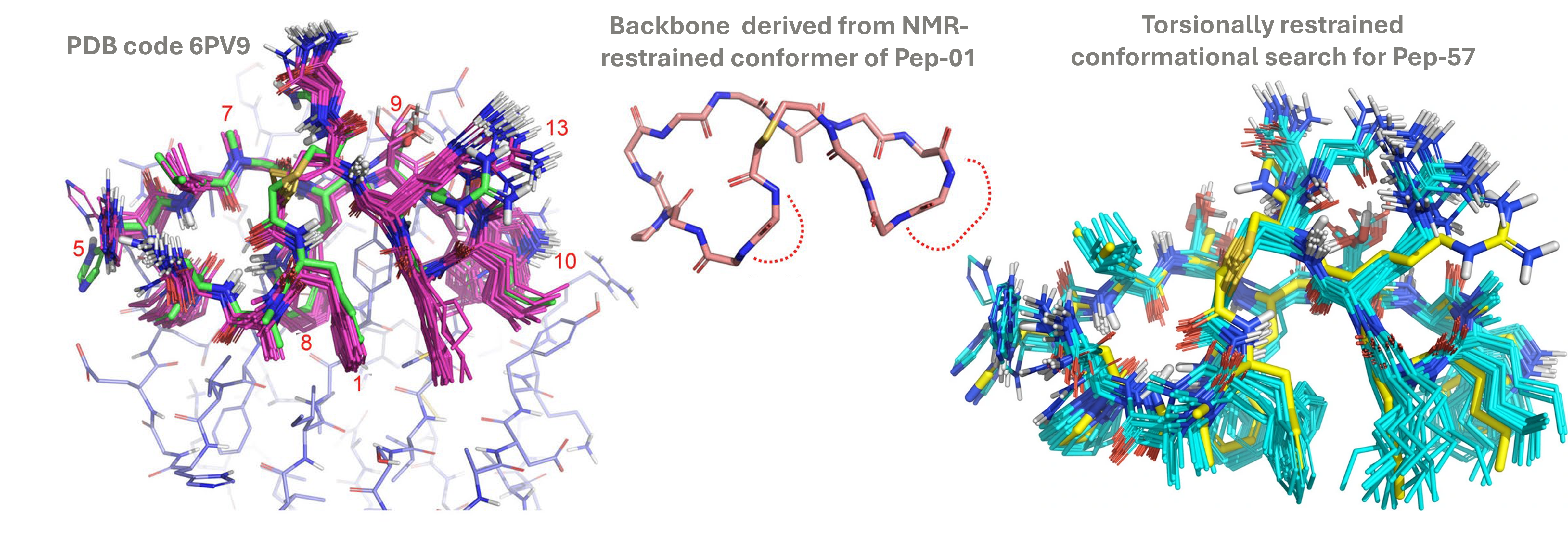
NMR restraints guide biologically relevant conformations
Lowest energy non-redundant conformers from NMR-restrained search of Pep-01 (magenta) superposed on its crystallographic pose (green) (left). The backbone derived from the lowest energy conformer (middle). Low energy conformers of Pep-57 (cyan), superimposed on its crystallographic pose (yellow) (right). Deposited structures were re-fitted using xGen to obtain best fits to the X-ray density, ensuring accurate comparisons.
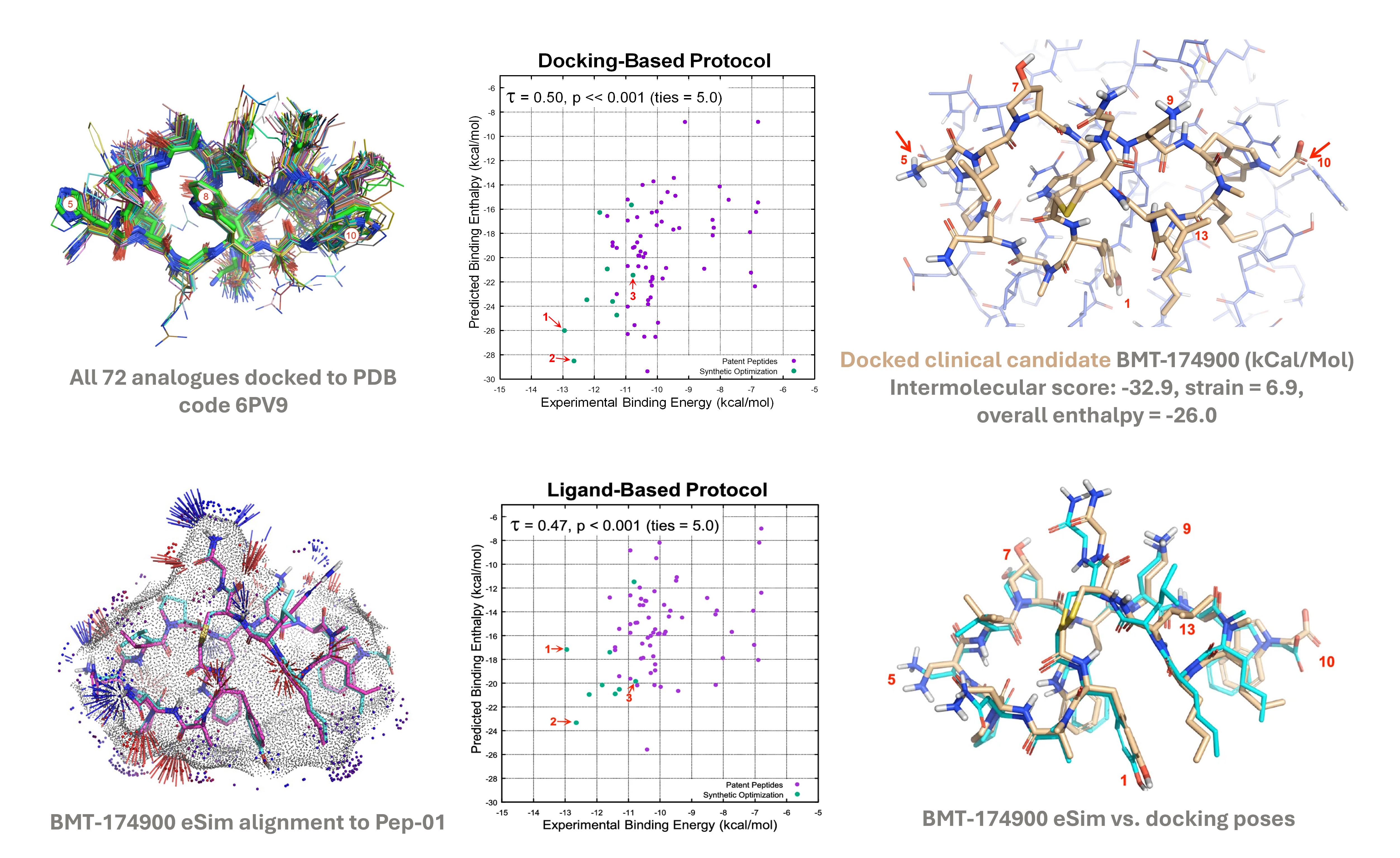
Docking/ligand-based ranking helps prioritise analogues
Docked poses of all analogues superimposed on the bound pose of Pep-01 (green) (top-left) and the predicted bound pose for the clinical candidate BMT-174900 (top-right). Optimal ligand-based alignment of BMT-174900 to Pep-01 for pose prediction (bottom-left) compared to its docked pose (bottom-right). Experimental vs predicted binding energies show synergy and both protocols correctly rank optimised peptides (green). The points 1, 2, and 3 correspond to three of the optimised peptides, BMT-174900, BMT- 153099, and BMT-139699, respectively (middle).
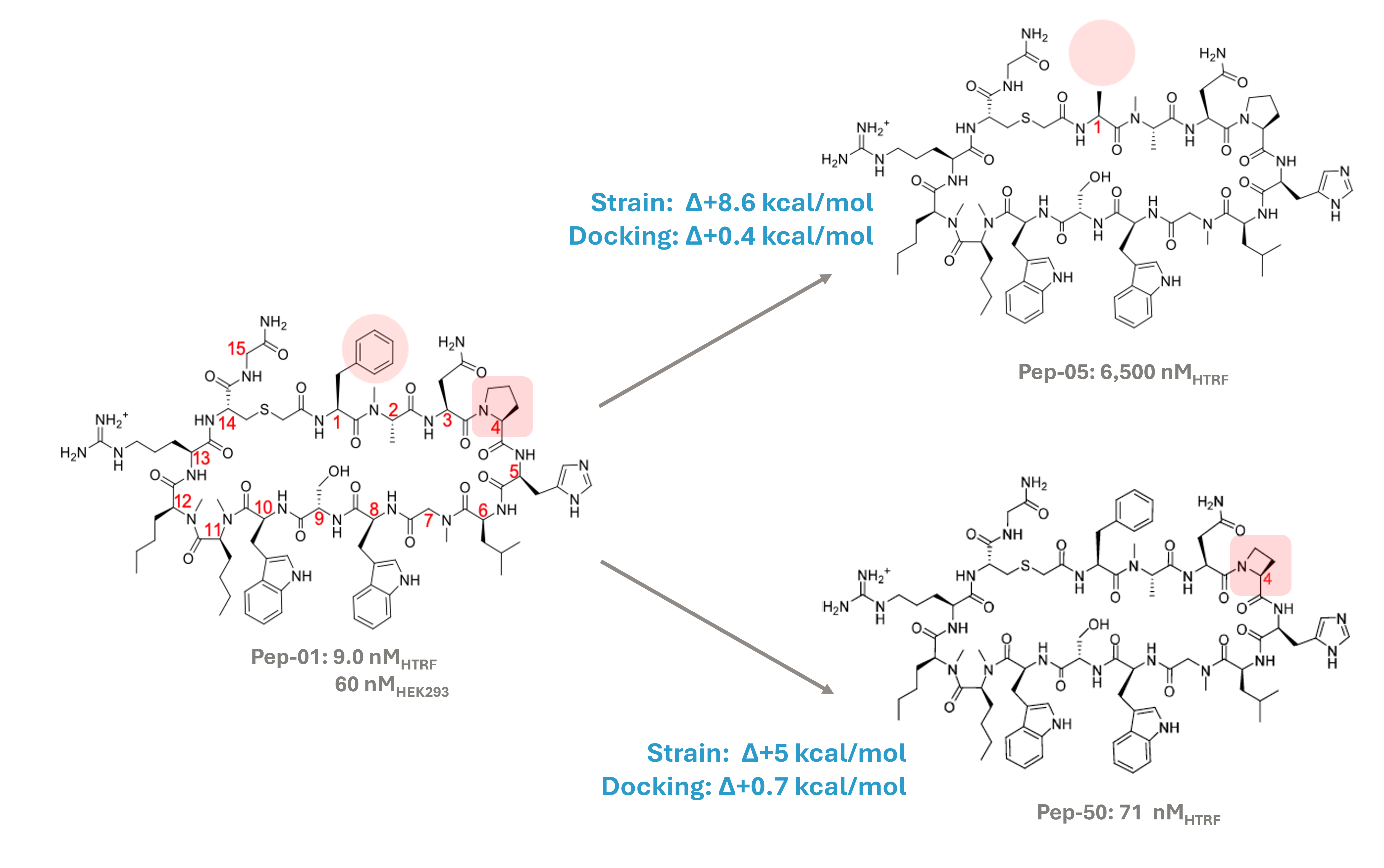
Ligand strain is a dominant predictive component
Substituting phenylalanine with alanine at position 1 in Pep-05 reduces activity by 3 log units despite a minor loss in intermolecular binding energy (<0.5 kcal/mol). However, this change increases macrocycle strain by ~8 kcal/mol. Conformational “locking” with rigid substituents significantly impacts strain. Similarly, deleting a single methylene from proline residue at position 4 minimally affects the interaction footprint (0.7 kcal/mol) but increases strain by 5 kcal/mol.
Conclusion
3D molecular modeling accelerates macrocycle lead optimisation with three key elements:
- A fast, rigorous and template-free method for macrocycle conformational search
- Effective use of biophysical data to constrain the conformational search and identify bioactive conformations
- An accurate model of ligand strain; small ligand modifications can cause large changes in strain, significantly impacting binding energy
References
- Miller MM, Mapelli C, Allen MP, et al US Patent 9:308
- Jain AN, Brueckner AC, Cleves AE, et al (2023) J Med Chem 66(3) https://doi.org/10.1021/acs.jmedchem.2c01744
- Cleves AE, Jain AN (2015) J Comput Aided Mol Des, 29 https://doi.org/10.1007/s10822-015-9846-3
- Cleves AE, Johnson SR, Jain AN (2019) J Comput Aided Mol Des 33(10) https://doi.org/10.1007/s10822-019-00236-6
This work: Jain AN, Brueckner, AC, Jorge, C, et al (2023) J Comput Aided Mol Des 37 https://doi.org/10.1007/s10822-023-00524-2