Does AI remove bias in drug discovery?
Like all humans, drug discovery scientists suffer from inherent biases that influence our decision making. Our intuition can sometimes be…

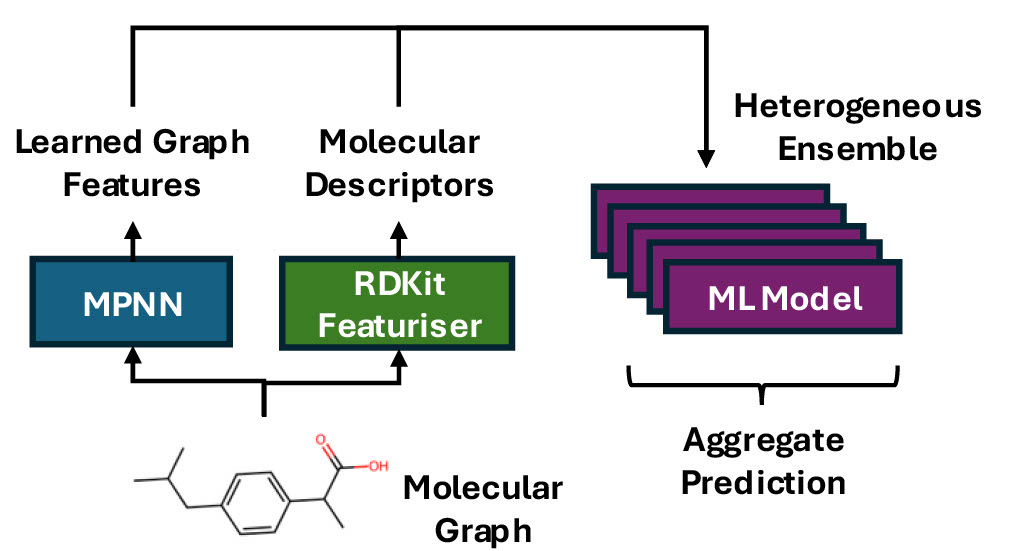
We explore a “best-of-both” approach to modelling molecular properties by combining learned molecular descriptors from a graph neural network (GNN) with general-purpose descriptors and a mixed ensemble of machine learning (ML) models.
We introduce a MetaModel framework to aggregate predictions from a diverse set of leading ML models. We present a featurisation scheme for combining task-specific GNN-derived features with conventional molecular descriptors.
We demonstrate that our framework outperforms the cutting-edge ChemProp model on all regression data sets tested and 6 of 9 classification data sets. We further show that including the GNN features derived from ChemProp boosts the ensemble model’s performance on several data sets where it otherwise would have underperformed. We conclude that to achieve optimal performance across a wide set of problems, it is vital to combine general-purpose descriptors with task-specific learned features and to use a diverse set of ML models to make the predictions.
Like all humans, drug discovery scientists suffer from inherent biases that influence our decision making. Our intuition can sometimes be…
If you’re using predictive models in your molecule design and optimisation, an accurate uncertainty estimate can be just as important…

What are neural networks? Neural networks (NNs) in various forms are very common nowadays, and specific architectures are used for…
