How can I model large molecules like macrocycles?
What comprises large molecules? When we talk about “large molecules,” we often think of biologics like monoclonal antibodies, proteins, and…

The similarity principle is one of the key concepts in drug design. It implies that making a series of minor changes to a molecule shouldn’t drastically affect how strongly it binds to its target. The challenge, however, is that the principle doesn’t tell us how to measure or define similarity between molecules in practice. Moreover, while local structural changes may preserve activity, this continuity does not necessarily hold over a long series of small modifications1.
Exceptions to this principle, where structurally similar compounds exhibit noticeably different activities, are referred to as activity cliffs. Mathematically, they are defined by the ratio of the difference in activity of two compounds to their distance of separation in a given chemical space2(we’ll speak more about chemical space later). In a nutshell, activity cliffs occur when structurally similar compounds targeting the same target exhibit large differences in potency. Due to their complexity, activity cliffs are difficult to model computationally and challenging to identify systematically3.
The most widely used approaches convert molecular structures into binary or count-based vectors, which are then compared using similarity metrics. The vectors are commonly known as molecular fingerprints and form the basis for many cheminformatics tools that enable rapid comparison of large compound libraries.
Fingerprints can be represented as:
Each type of molecular fingerprint comes with its own advantages and limitations, so selecting the most appropriate one for a specific task should be guided by benchmarking studies tailored to the problem at hand1.
Below, we’ll cover some of the most common fingerprint types. Until now, we have focused on 2D fingerprints; however, it is important to note that 3D fingerprints (e.g., 3D pharmacophore fingerprints) are also widely used, though they heavily depend on the molecular conformation. A detailed discussion of these methods, however, lies beyond the scope of this blog.
Path-based molecular fingerprints are among the oldest types used in cheminformatics. They analyse linear paths through molecular graphs – sequences of atoms and bonds, up to a predefined maximum length (typically 5 to 7 atoms). Each unique path is hashed into a position in a fixed-size bit vector, commonly 1024 bits4. These fingerprints are widely used for molecular similarity searching and substructure matching due to their simplicity and computational efficiency.
A notable subcategory of path-based fingerprints is 2D pharmacophore fingerprints, where atoms are annotated with pharmacophoric features (such as hydrogen bond donors, acceptors, or aromatic rings), and the paths between these pharmacophore points are encoded.
This approach creates fingerprints based on atom pairs within molecules. Each pair is defined by the atom types, along with the topological distance (the number of bonds between them). These are typically encoded as triplets: (atom type 1, atom type 2, bond distance).
To generate a fixed-length fingerprint, each unique atom pair is hashed into a numerical value and mapped to an index within a fixed-size vector. Since the number of possible atom pairs is vast, this mapping is many-to-one, meaning that different pairs can hash to the same index—a phenomenon known as a hash collision. For each pair, the corresponding index is either set to 1 (in a binary fingerprint) or incremented (in a count-based fingerprint). The result is a compact yet informative representation of a molecule’s medium-range structural features, well-suited for rapid similarity comparisons5.
This approach captures the local atomic environment around each atom in a molecule. It iteratively gathers information about neighbouring atoms up to a certain radius (number of bonds), effectively encoding atom-centered substructures. These neighbourhoods are then hashed into identifiers, which represent unique structural patterns. As with atom pairs, the hashed identifiers are mapped into a fixed-length vector using a hash function that assigns each feature to a specific index.
Due to the extremely large number of possible substructures, hash collisions can occur where multiple patterns map to the same index6. Circular fingerprints are particularly useful for separating active compounds from inactives, where they may have similar physical properties in a virtual screen.
The Tanimoto coefficient is the most common method of quantifying molecular similarity, particularly with binary fingerprints. It measures the overlap between two fingerprint vectors by comparing the number of shared features to the total number of unique features. The result is a similarity score ranging from 0 (no similarity) to 1 (identical).
While this provides a rigorous, numerical measure of similarity, it is often useful to complement it with a visual representation. This is typically done by projecting high-dimensional molecular descriptors into a 2D chemical space using dimensionality reduction techniques such as PCA, t-SNE, or UMAP (see the image below). In this space, similar compounds tend to cluster together, allowing researchers to visually inspect relationships that might not be obvious from numerical scores alone. Thus, similarity can be both calculated and seen, providing complementary perspectives on molecular relationships.
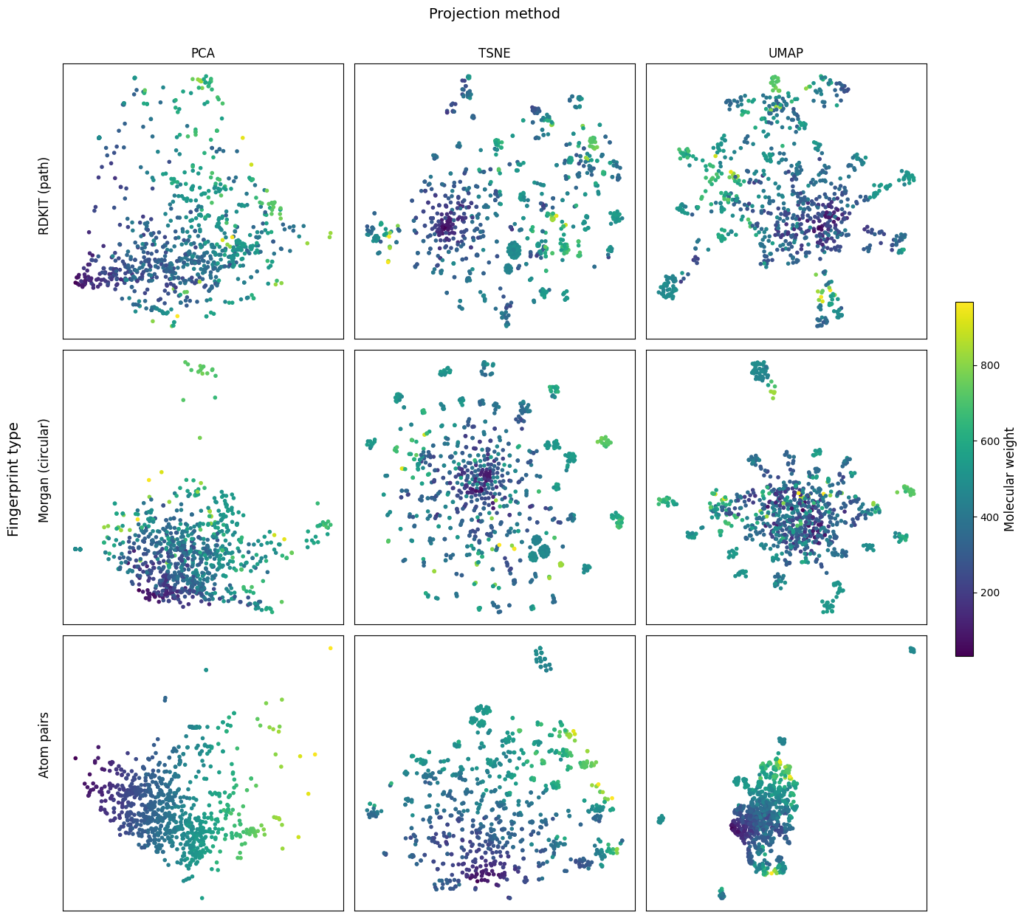
When analysing similarity, how different is meaningfully different? In other words, what size difference in score reflects a meaningful difference in biological or chemical behaviour?
For example, a Tanimoto similarity of 0.85 versus 0.75 may appear close numerically, but depending on the fingerprint type and context (e.g., target class, assay type), this difference might correspond to a substantial change in activity or none at all.
To assess statistical significance, researchers often use benchmark datasets that correlate similarity scores to known properties like bioactivity. By comparing distributions of similarities between active–active, active–inactive, and random compound pairs, it’s possible to define thresholds where similarity differences become statistically meaningful.
Molecular similarity is a fundamental concept in drug discovery: the greater the resemblance between two molecules, the more likely they are to exhibit similar biological activity. Computational techniques are commonly used to convert molecular structures into digital fingerprints—simplified representations of molecular features—that can be compared through various algorithms. Numerous fingerprinting methods exist, but no single approach is universally applicable, as molecules that appear nearly identical in chemical structure or computational chemical space may behave differently in biological systems. Therefore, determining true similarity between compounds is highly context-dependent.
Mario is a Principal Scientist at Optibrium, where he applies expertise in quantum chemistry, conformational analysis, and molecular modeling to support drug discovery research. With a PhD in Natural Sciences from Tallinn University of Technology, his academic work focused on computational studies of macrocyclic molecules. Mario has authored numerous publications and has extensive experience in chemical education and advanced spectroscopy.

What comprises large molecules? When we talk about “large molecules,” we often think of biologics like monoclonal antibodies, proteins, and…
Introduction 3D molecular modelling plays a vital role in modern drug discovery, offering powerful applications to streamline research, reduce costs,…

In this paper, we describe an extended benchmark for non-cognate docking of macrocyclic ligands, and the superior performance of Surflex™-Dock…